Novel TNXB Variants in Two Italian Patients with Classical-Like Ehlers-Danlos Syndrome

 , ,
, ,  ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation and Next Generation Sequencing Analysis
2.2. Sanger Sequencing.
2.3. Analysis of the TNXA/TNXB Homology Region
2.4. Multiplex Ligation-Dependent Probe Amplification (MLPA) and Quantitative PCR (qPCR) Analysis
2.5. Chromosome Microarrays Analysis (CMA)
2.6. Conservation of the Variant.
2.7. Total RNA Analysis.
2.8. Variant Designation.
3. Results
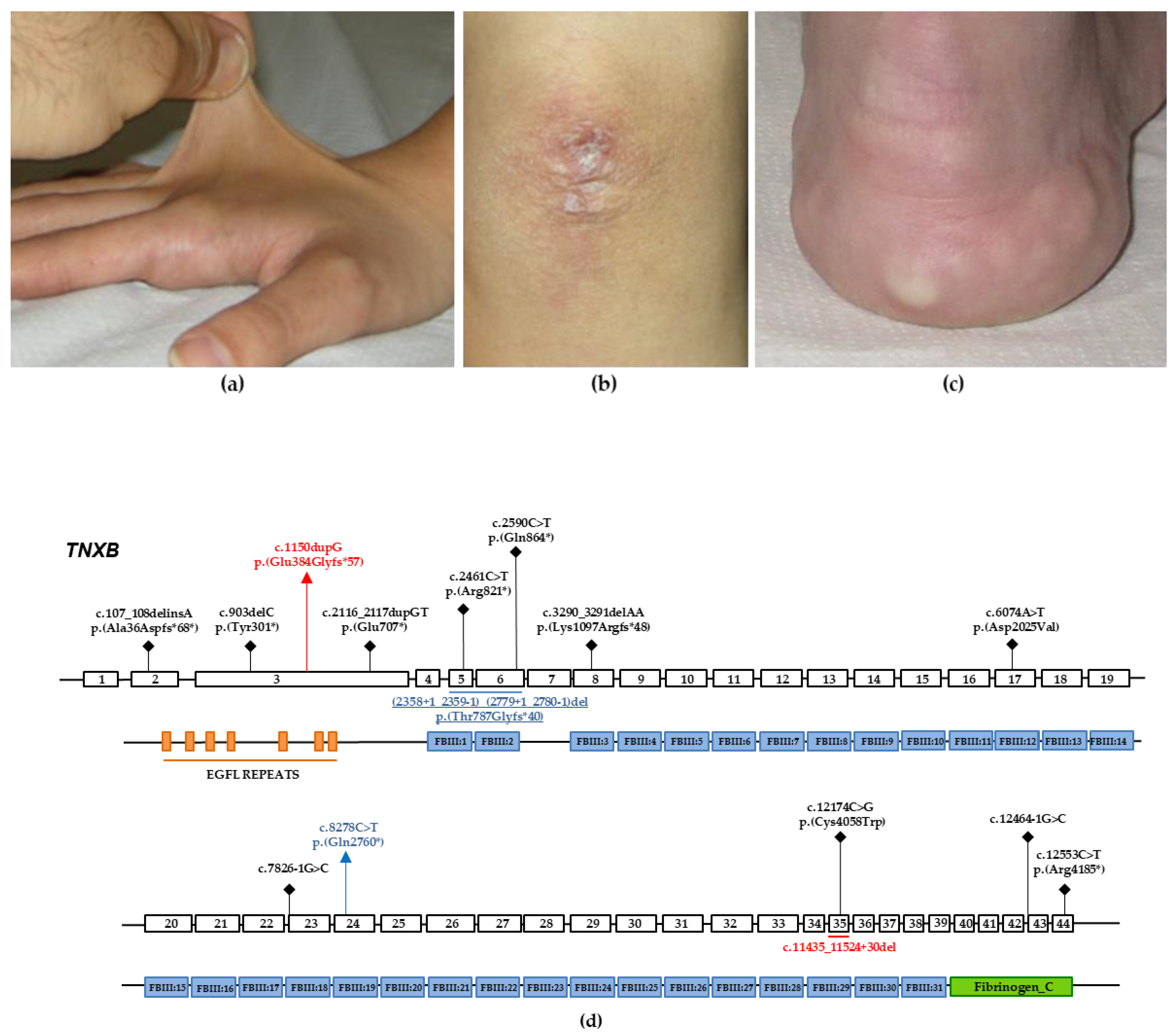
3.1. Case Report: Individual 1
3.2. Case Report: Individual 2
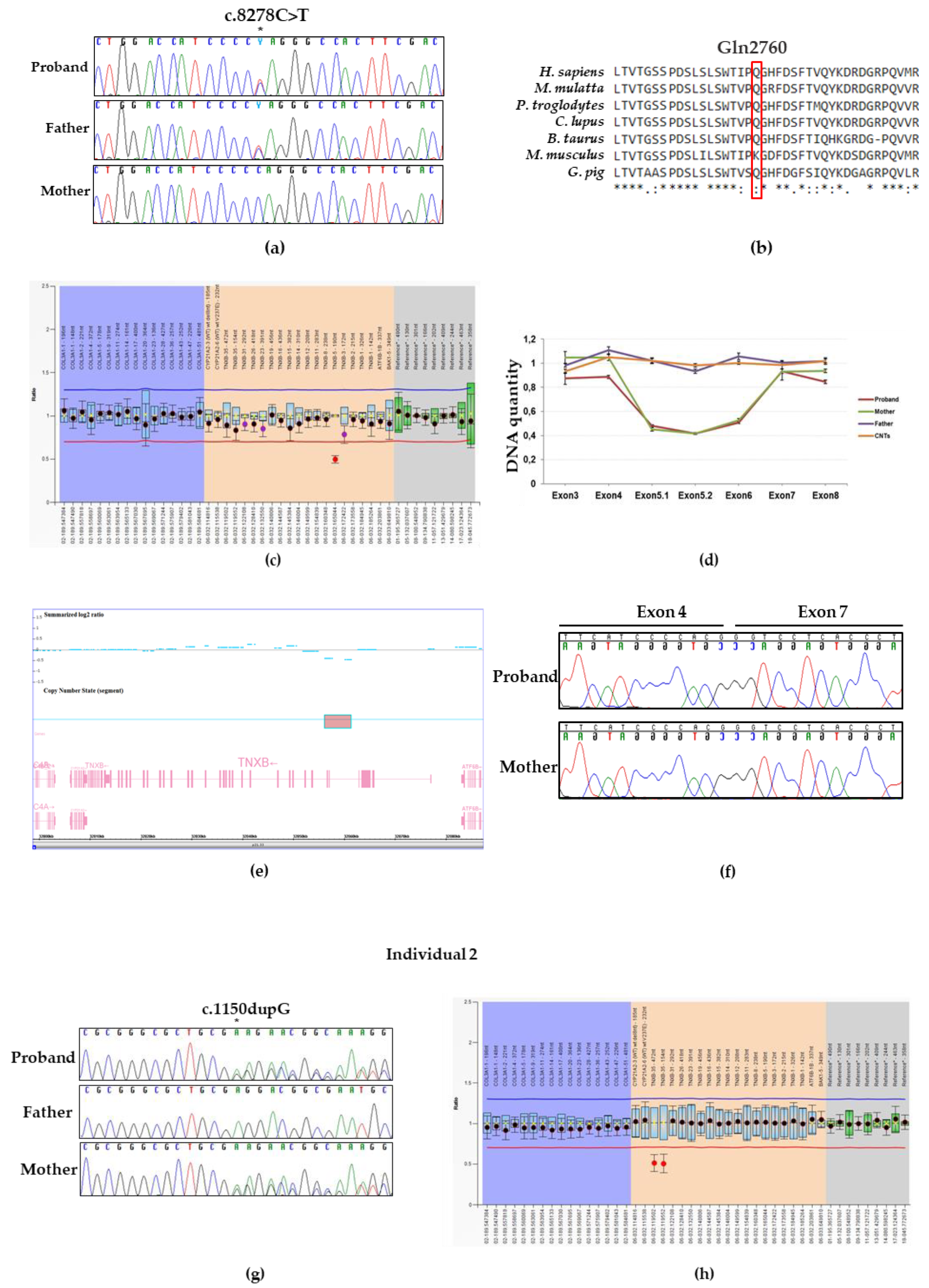
3.3. Molecular Findings: Individual 1
3.4. Molecular Findings: Individual 2
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [PubMed]
- Ritelli, M.; Cinquina, V.; Venturini, M.; Pezzaioli, L.; Formenti, A.M.; Chiarelli, N.; Colombi, M. Expanding the Clinical and Mutational Spectrum of Recessive AEBP1-Related Classical-Like Ehlers-Danlos Syndrome. Genes (Basel) 2019, 10, 135. [Google Scholar] [CrossRef] [PubMed]
- Syx, D.; De Wandele, I.; Symoens, S.; De Rycke, R.; Hougrand, O.; Voermans, N.; De Paepe, A.; Malfait, F. Bi-allelic AEBP1 mutations in two patients with Ehlers-Danlos syndrome. Hum. Mol. Genet. 2019, 28, 1853–1864. [Google Scholar] [CrossRef] [PubMed]
- Burch, G.H.; Gong, Y.; Liu, W.; Dettman, R.W.; Curry, C.J.; Smith, L.; Miller, W.L.; Bristow, J. Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nat. Genet. 1997, 17, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, J.; Zweers, M.C.; Steijlen, P.M.; Dean, W.B.; Taylor, G.; van Vlijmen, I.M.; van Haren, B.; Miller, W.L.; Bristow, J. A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N. Engl. J. Med. 2001, 345, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Brady, A.F.; Demirdas, S.; Fournel-Gigleux, S.; Ghali, N.; Giunta, C.; Kapferer-Seebacher, I.; Kosho, T.; Mendoza-Londono, R.; Pope, M.F.; Rohrbach, M.; et al. The Ehlers-Danlos syndromes, rare types. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 70–115. [Google Scholar] [CrossRef] [PubMed]
- Demirdas, S.; Dulfer, E.; Robert, L.; Kempers, M.; van Beek, D.; Micha, D.; van Engelen, B.G.; Hamel, B.; Schalkwijk, J.; Loeys, B.; et al. Recognizing the tenascin-X deficient type of Ehlers-Danlos syndrome: A cross-sectional study in 17 patients. Clin. Genet. 2017, 91, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Perritt, A.F.; Morissette, R.; Dreiling, J.L.; Bohn, M.F.; Mallappa, A.; Xu, Z.; Quezado, M.; Merke, D.P. Ehlers-Danlos Syndrome Caused by Biallelic TNXB Variants in Patients with Congenital Adrenal Hyperplasia. Hum. Mutat. 2016, 37, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Merke, D.P.; Chen, W.; Morissette, R.; Xu, Z.; Van Ryzin, C.; Sachdev, V.; Hannoush, H.; Shanbhag, S.M.; Acevedo, A.T.; Nishitani, M.; et al. Tenascin-X haploinsufficiency associated with Ehlers-Danlos syndrome in patients with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2013, 98, E379–E387. [Google Scholar] [CrossRef] [PubMed]
- Morissette, R.; Chen, W.; Perritt, A.F.; Dreiling, J.L.; Arai, A.E.; Sachdev, V.; Hannoush, H.; Mallappa, A.; Xu, Z.; McDonnell, N.B.; et al. Broadening the Spectrum of Ehlers Danlos Syndrome in Patients With Congenital Adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2015, 100, E1143–E1152. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Copetti, M.; Morlino, S.; Colombi, M.; Grammatico, P.; Fontana, A.; Castori, M. Severity classes in adults with hypermobile Ehlers-Danlos syndrome/hypermobility spectrum disorders: A pilot study of 105 Italian patients. Rheumatology (Oxford) 2019, 58, 1722–1730. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, C.S.; Butler, M.G. Mutation in TNXB gene causes moderate to severe Ehlers-Danlos syndrome. World J. Med. Genet. 2016, 6, 17–21. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Primers | Sequence | Tm (°C) | Size (bp) |
|---|---|---|---|
| TNXB-LongPCR-F | GTCTCTGCCCTGGGAATGA | 60 | 4900 |
| TNXB-LongPCR-R | TGTAAACACAGTGCTGCGA | ||
| TNXB frag1 For | GGCCAAGCCTGGAAGATAAA | 60 | 662 |
| TNXB frag1 Rev | GATTGGAGACAGAAGCACAC | ||
| TNXB frag2 For | CCAGGGAGAGAGGATGGAT | 60 | 671 |
| TNXB frag2 Rev | GTCCCCAGGAATGGAAGT | ||
| TNXB frag3 For | GACCTAGTGCCTCAGCCA | 60 | 733 |
| TNXB frag3 Rev | GGCTCTCTCTACTCCGTG | ||
| TNXB frag4 For | ATGGGTGGGAGTTGAGAG | 60 | 727 |
| TNXB frag4 Rev | TGGAAGCTGAGCAGGTAG | ||
| TNXB frag5 For | TCTCCTCTTCCTGCTTTCCC | 60 | 643 |
| TNXB frag5 Rev | CCCCATCAGTCTCCATGTC | ||
| TNXB frag6 For | CAGGACCAGCACCATCTT | 60 | 741 |
| TNXB frag6 Rev | TTGAGGTTGGCGTAGTGG | ||
| TNXB frag7 For | GCTGTCTCCTACCGAGGG | 60 | 621 |
| TNXB frag7 Rev | GCAGAGAAGGCTTCCTCC | ||
| TNXB_EX3Fw | GGTTGCAGTCAGAGGGGGCG | 58 | 300 |
| TNXB_EX3Rv | GCCCGCGACCTCTACAGTCG | ||
| TNXB_EX35Fw | GGGAGCCTCAGAGTGTGC | 58 | 480 |
| TNXB_EX35Rv | TCAGCCCCTGGAGTTTCTG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Micale, L.; Guarnieri, V.; Augello, B.; Palumbo, O.; Agolini, E.; Sofia, V.M.; Mazza, T.; Novelli, A.; Carella, M.; Castori, M. Novel TNXB Variants in Two Italian Patients with Classical-Like Ehlers-Danlos Syndrome. Genes 2019, 10, 967. https://doi.org/10.3390/genes10120967
Micale L, Guarnieri V, Augello B, Palumbo O, Agolini E, Sofia VM, Mazza T, Novelli A, Carella M, Castori M. Novel TNXB Variants in Two Italian Patients with Classical-Like Ehlers-Danlos Syndrome. Genes. 2019; 10(12):967. https://doi.org/10.3390/genes10120967
Chicago/Turabian StyleMicale, Lucia, Vito Guarnieri, Bartolomeo Augello, Orazio Palumbo, Emanuele Agolini, Valentina Maria Sofia, Tommaso Mazza, Antonio Novelli, Massimo Carella, and Marco Castori. 2019. "Novel TNXB Variants in Two Italian Patients with Classical-Like Ehlers-Danlos Syndrome" Genes 10, no. 12: 967. https://doi.org/10.3390/genes10120967
APA StyleMicale, L., Guarnieri, V., Augello, B., Palumbo, O., Agolini, E., Sofia, V. M., Mazza, T., Novelli, A., Carella, M., & Castori, M. (2019). Novel TNXB Variants in Two Italian Patients with Classical-Like Ehlers-Danlos Syndrome. Genes, 10(12), 967. https://doi.org/10.3390/genes10120967