Transcriptome-Wide Analysis Reveals Key DEGs in Flower Color Regulation of Hosta plantaginea (Lam.) Aschers


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
2.2. RNA Extraction and Sequencing
2.3. Transcriptome Assembly
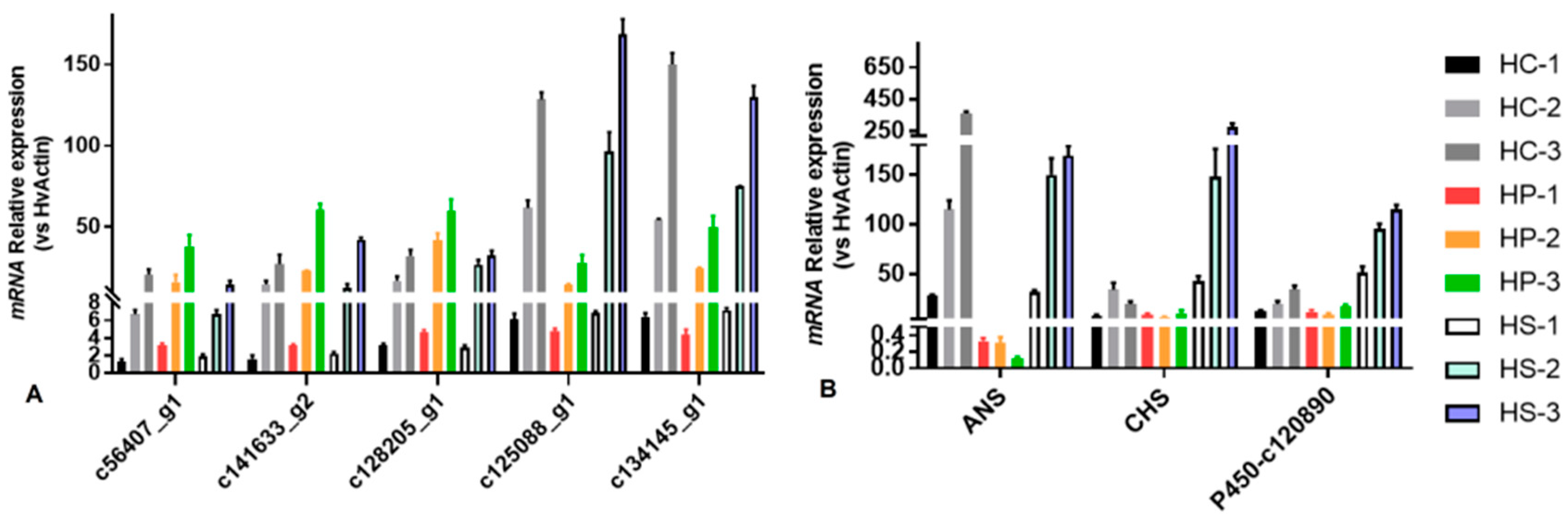
2.4. Validation of Gene Expression with qRT-PCR
2.5. Gene Ontology Annotation
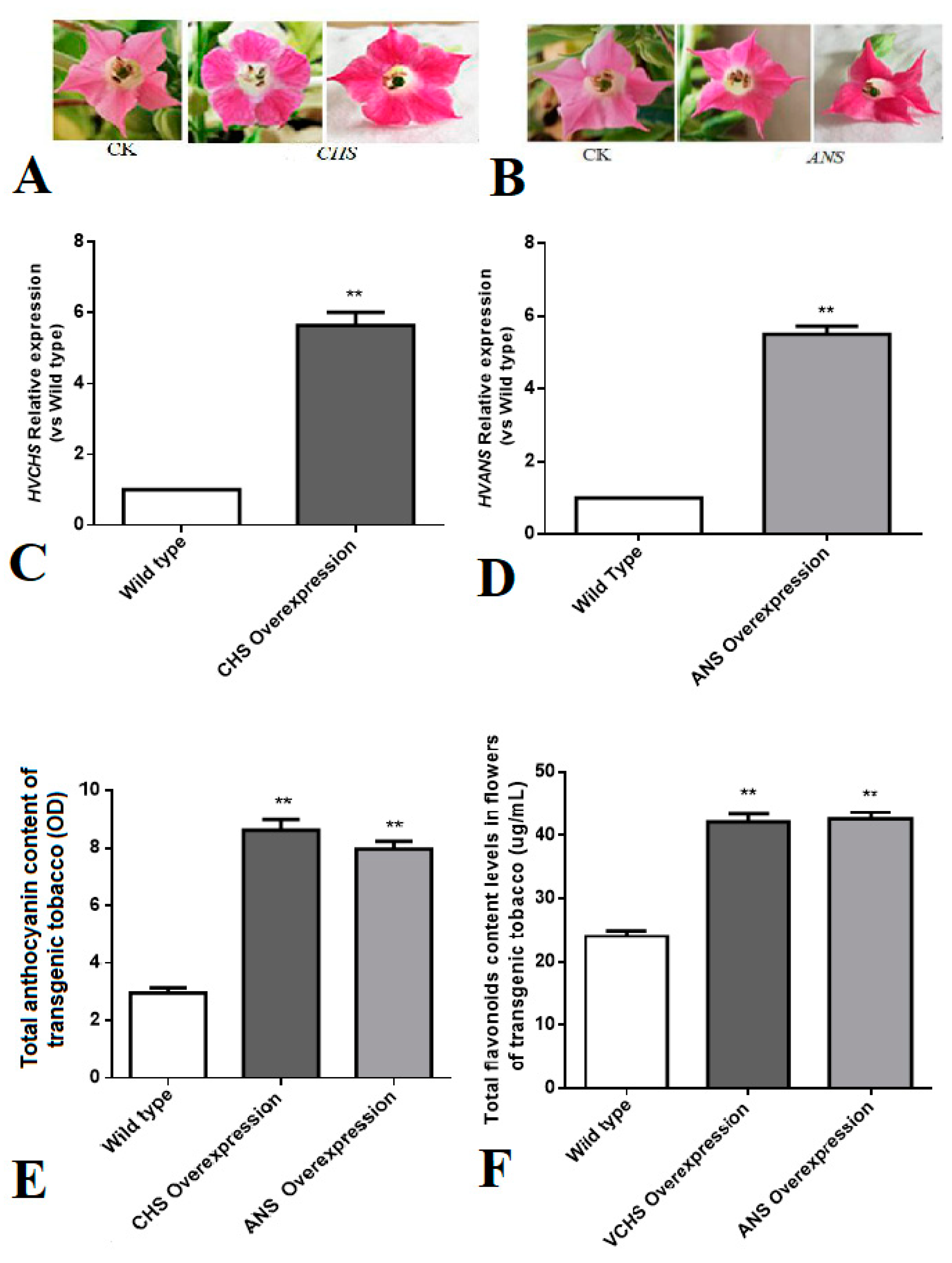
2.6. Investigation of Flower Color Regulation by CHS and ANS Genes in Tobacco
3. Results
3.1. Flower Colors of HPA at Different Developmental Stages
3.2. Transcriptome Sequencing and Assembly
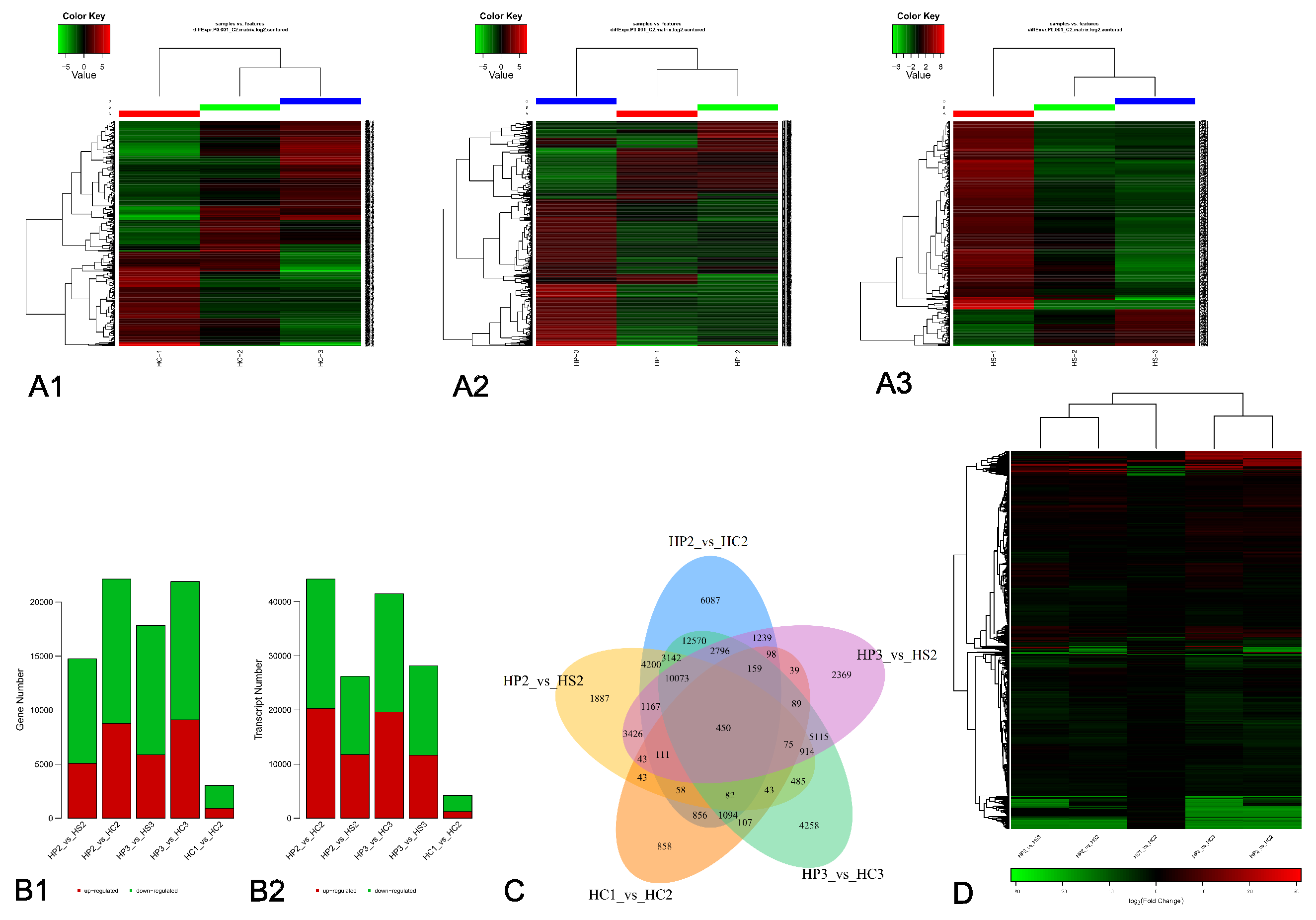
3.3. Analyses of DEGs
3.4. Validation of DEGs by qRT-PCR
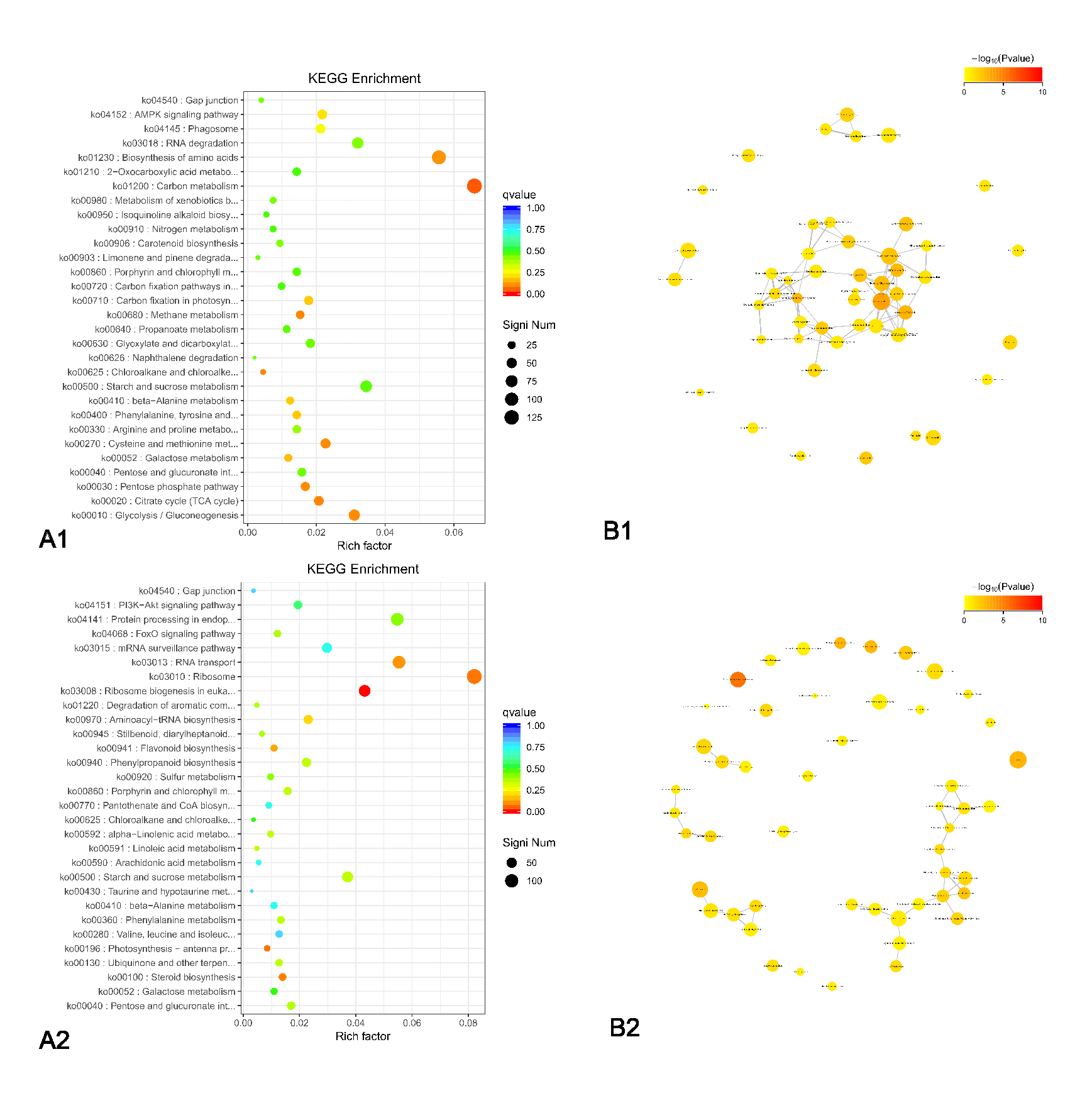
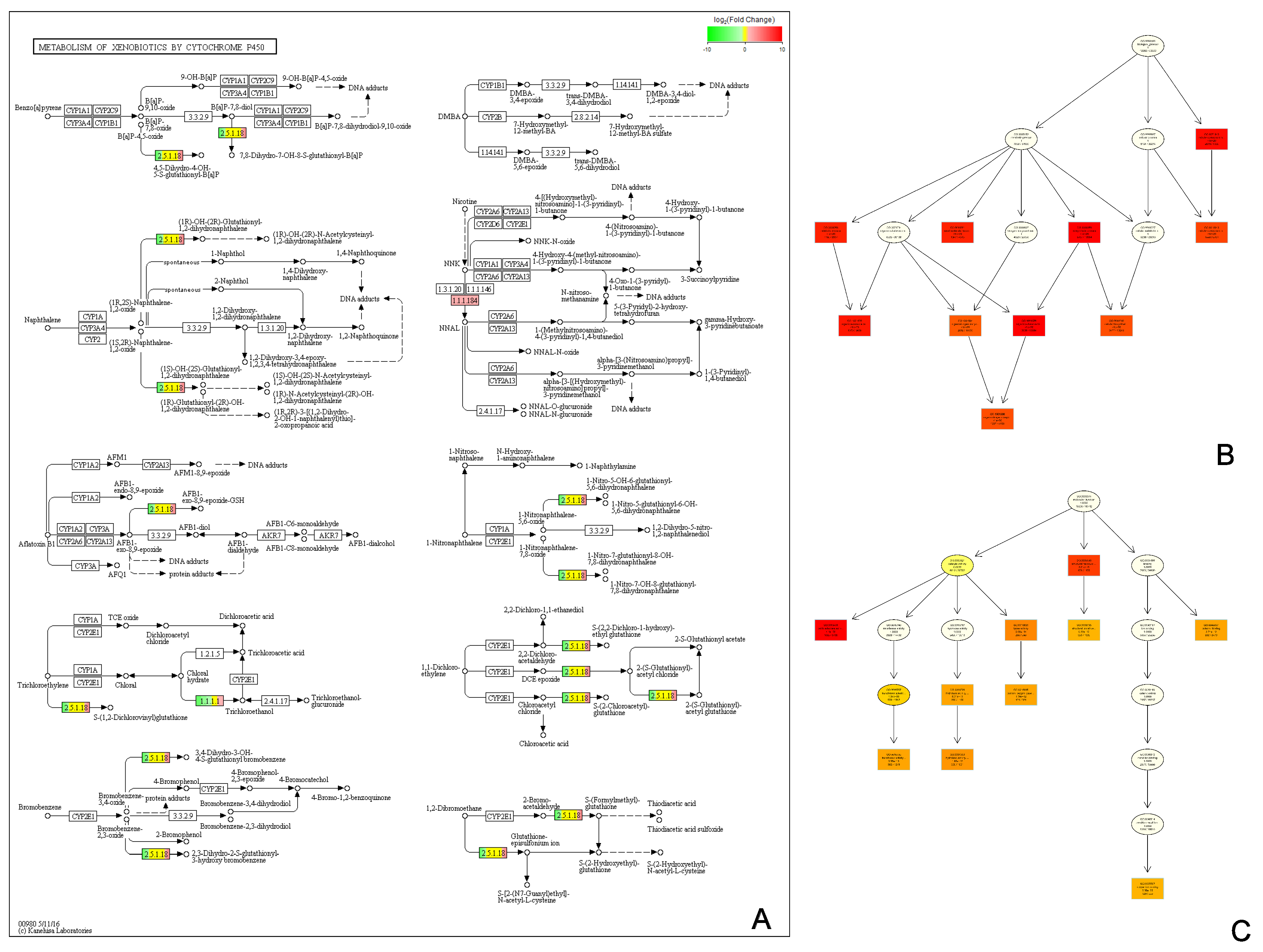
3.5. Pathway and Functional Analysis of DEGs
3.6. Study on Flower Color Regulation of CHS and ANS Genes in Tobacco
4. Discussions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Availability of Data and Materials
Abbreviations
| HPA | Hosta plantaginea (Lam.) Aschers or H. plantaginea (Lam.) A. |
| HC | Hosta Cathayana or H. Cathayana |
| HP | Hosta plantaginea or H. plantaginea |
| HS | Hosta plantaginea ‘Summer Fragrance’ or H. plantaginea ‘Summer Fragrance’ |
| DEGs | differentially expressed genes |
| CHS | Chalcone synthase |
| ANS | Anthocyanidin synthase |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KOG | Eukaryotic Orthologous Groups of protein |
| qRT-PCR | quantitative real-time PCR |
| UFGT | UDP-glucose:flavonoid-3-o-glucosyltransferase |
| DAG | Directed Acyclic Graph |
References
- Liu, J.; Zhao, C.H.; Liu, X.R.; Xi, Y.Z.; Zhang, Y.L. Pollen morphology of Hosta Tratt. in China and its taxonomic significance. Plant Syst. Evol. 2011, 294, 99–107. [Google Scholar] [CrossRef]
- Brouillard, R. The in vivo expression of anthocyanin colour in plants. Phytochem. Rev. 1983, 22, 1311–1323. [Google Scholar] [CrossRef]
- Bloor, S.J.; Falshaw, R. Covalently linked anthocyanin-flavonol pigments from blue Agapanthus flowers. Phytochemistry 2000, 53, 575–579. [Google Scholar] [CrossRef]
- Tanaka, Y.; Tsuda, S.; Kusumi, T. Metabolic Engineering to Modify Flower Color. Plant Cell Physiol. 1998, 39, 1119–1126. [Google Scholar] [CrossRef]
- Tanaka, Y.; Ohmiya, A. Seeing is believing: Engineering anthocyanin and carotenoid biosynthetic pathways. Curr. Opin. Biotechnol. 2008, 19, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Forkmann, G. Flavonoids as Flower Pigments: The Formation of the Natural Spectrum and its Extension by Genetic Engineering. Plant Breed. 1991, 106, 1–26. [Google Scholar] [CrossRef]
- Aida, R.; Yoshida, K.; Kondo, T.; Kishimoto, S.; Shibata, M. Copigmentation gives bluer flowers on transgenic torenia plants with the antisense dihydroflavonol-4-reductase gene. Plant Sci. 2000, 160, 49–56. [Google Scholar] [CrossRef]
- Grotewold, E. The genetics and biochemistry of floral pigments. Annu. Rev. Plant Biol. 2006, 57, 761–780. [Google Scholar] [CrossRef]
- Yukihisa, K.; Masako, F.-M.; Yuko, F.; Filippa, B.; Timothy, A.H.; Mirko, K.; Noriko, N.; Keiko, Y.-S.; Junichi, T.; Alix, P. Engineering of the Rose Flavonoid Biosynthetic Pathway Successfully Generated Blue-Hued Flowers Accumulating Delphinidin. Plant Cell Physiol. 2007, 48, 1589–1600. [Google Scholar]
- Tanaka, Y.; Brugliera, F. Flower colour and cytochromes P450. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368. [Google Scholar] [CrossRef] [Green Version]
- Falcone Ferreyra, M.L.; Emiliani, J.; Rodriguez, E.J.; Campos-Bermudez, V.A.; Grotewold, E.; Casati, P. The identification of maize and Arabidopsis type I flavone synthases links flavones with hormones and biotic interactions. Plant Physiol. 2015, 169, 1090–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buer, C.S.; Djordjevic, M.A. Architectural phenotypes in the transparent testa mutants of Arabidopsis thaliana. J. Exp. Bot. 2009, 60, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Buer, C.S.; Muday, G.K.; Djordjevic, M.A. Flavonoids Are Differentially Taken up and Transported Long Distances in Arabidopsis. Plant Physiol. 2007, 145, 478–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manfred, G.G.; Brian, J.H.; Moran, Y.; Joshua, Z.L.; Dawn, A.T.; Ido, A.; Xian, A.; Lin, F.; Raktima, R.; Qiandong, Z. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Wu, P.-Y.; Phan, J.H.; Wang, M.D. An Approach for Assessing RNA-seq Quantification Algorithms in Replication Studies. In Proceedings of the 2013 IEEE International Workshop on Genomic Signal Processing and Statistics, Houston, TX, USA, 17–19 November 2013; Volume 2013, pp. 15–18. [Google Scholar]
- Borodina, T.; Adjaye, J.; Sultan, M. A strand-specific library preparation protocol for RNA sequencing. Methods Enzymol. 2011, 500, 79–98. [Google Scholar]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, 182–185. [Google Scholar] [CrossRef] [Green Version]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Götz, S. Blast2GO: A Comprehensive Suite for Functional Analysis in Plant Genomics. Int. J. Plant Genom. 2008, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dan, W. Clonging of the Grazing-Tolerant Ralated Gene of Medicago Falcata and Transformation in Tobacaco. Ph.D. Thesis, Inner Mongolia Agricultural University, Hohhot, China, 2015. [Google Scholar]
- Hui, L. Cloning and Functional Analysis of ZmWRKY25 Gene from Zea mays. Ph.D. Thesis, Jilin Agricultural University, Changchun, China, 2017. [Google Scholar]
- Wang, Y.; Dou, Y.; Wang, R.; Guan, X.; Hu, Z.; Zheng, J. Molecular characterization and functional analysis of chalcone synthase from Syringa oblata Lindl. in the flavonoid biosynthetic pathway. Gene 2017, 635, 16–23. [Google Scholar] [CrossRef] [PubMed]
- GandíaHerrero, F.; GarcíaCarmona, F. Biosynthesis of betalains: Yellow and violet plant pigments. Trends Plant Sci. 2013, 18, 334–343. [Google Scholar] [CrossRef]
- Zhao, D.; Tao, J. Recent advances on the development and regulation of flower color in ornamental plants. Front. Plant Sci. 2015, 6, 216. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.X.; Zhang, J.H.; Zhang, H.G.; Xue, P.F. Studies on chemical constituents from Hosta plantaginea (Lam.) Aschers, a mongolia medicine. Chin. Pharm. J. 2009, 4, 733–735. [Google Scholar]
- Peifeng, X.; Jinhua, Z.; Hongxia, X.; Xiaojuan, L.; Guoying, W. Advances in studies on chemical constituents and pharmacological effects of hosta. Chin. Herb. Med. 2011, 34, 647–651. [Google Scholar]
- Liu, N. Analysis of Hosta and Its Relationship with Flower Color. Ph.D. Thesis, Institute of botany, Chinese Academy of Sciences, Beijing, China, 2012. [Google Scholar]
- Tan, J.; Wang, M.; Tu, L.; Nie, Y.; Lin, Y.; Zhang, X. The Flavonoid Pathway Regulates the Petal Colors of Cotton Flower. PLoS ONE 2013, 8, e72364. [Google Scholar] [CrossRef] [Green Version]
- Schuler, M.A. The Role of Cytochrome P450 Monooxygenases in Plant-Insect Interactions. Plant Physiol. 1996, 112, 1411–1419. [Google Scholar] [CrossRef] [Green Version]
- Nan, Z.; Tootle, T.L.; Glazebrook, J. Arabidopsis PAD3, a Gene Required for Camalexin Biosynthesis, Encodes a Putative Cytochrome P450 Monooxygenase. Plant Cell 1999, 11, 2419–2428. [Google Scholar]
- Holton, T.A.; Brugliera, F.; Lester, D.R.; Tanaka, Y.; Hyland, C.D.; Menting, J.G.T.; Lu, C.-Y.; Farcy, E.; Stevenson, T.W.; Cornish, E.C. Cloning and expression of cytochrome P450 genes controlling flower colour. Nature 1993, 366, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Stotz, G.; Vlaming, P.; Wiering, H.; Schram, A.W.; Forkmann, G. Genetic and biochemical studies on flavonoid 3′-hydroxylation in flowers ofPetunia hybrida. Theor. Appl. Genet. 1985, 70, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Ning, Z.; Zongli, H.; Xuqing, C.; Xiaoshu, H.; Yong, L.; Guoping, C. Analysis of Metabolic Pathway and Establishment of Regulating Model of Anthocyanin Synthesis. Chin. Biotechnol. 2008, 28, 97–105. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Sequence | %GC | Sequence Length (bp) |
|---|---|---|---|
| HC-1 | 48,527,189 | 47.5 | 125 |
| HC-2 | 48,085,043 | 48 | 125 |
| HC-3 | 48,250,194 | 48.5 | 125 |
| HP-1 | 44,116,660 | 47 | 125 |
| HP-2 | 44,512,366 | 48 | 125 |
| HP-3 | 42,504,784 | 48 | 125 |
| HS-1 | 43,869,339 | 47 | 125 |
| HS-2 | 44,195,154 | 47 | 125 |
| HS-3 | 44,053,222 | 47 | 125 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Sui, C.; Wang, Y.; Liu, S.; Liu, H.; Zhang, Z.; Liu, H. Transcriptome-Wide Analysis Reveals Key DEGs in Flower Color Regulation of Hosta plantaginea (Lam.) Aschers. Genes 2020, 11, 31. https://doi.org/10.3390/genes11010031
Zhang J, Sui C, Wang Y, Liu S, Liu H, Zhang Z, Liu H. Transcriptome-Wide Analysis Reveals Key DEGs in Flower Color Regulation of Hosta plantaginea (Lam.) Aschers. Genes. 2020; 11(1):31. https://doi.org/10.3390/genes11010031
Chicago/Turabian StyleZhang, Jingying, Changhai Sui, Yanli Wang, Shuying Liu, Huimin Liu, Zhengren Zhang, and Hongzhang Liu. 2020. "Transcriptome-Wide Analysis Reveals Key DEGs in Flower Color Regulation of Hosta plantaginea (Lam.) Aschers" Genes 11, no. 1: 31. https://doi.org/10.3390/genes11010031
APA StyleZhang, J., Sui, C., Wang, Y., Liu, S., Liu, H., Zhang, Z., & Liu, H. (2020). Transcriptome-Wide Analysis Reveals Key DEGs in Flower Color Regulation of Hosta plantaginea (Lam.) Aschers. Genes, 11(1), 31. https://doi.org/10.3390/genes11010031