Landscape Genomics of a Widely Distributed Snake, Dolichophis caspius (Gmelin, 1789) across Eastern Europe and Western Asia

 , , , , ,
, , , , ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
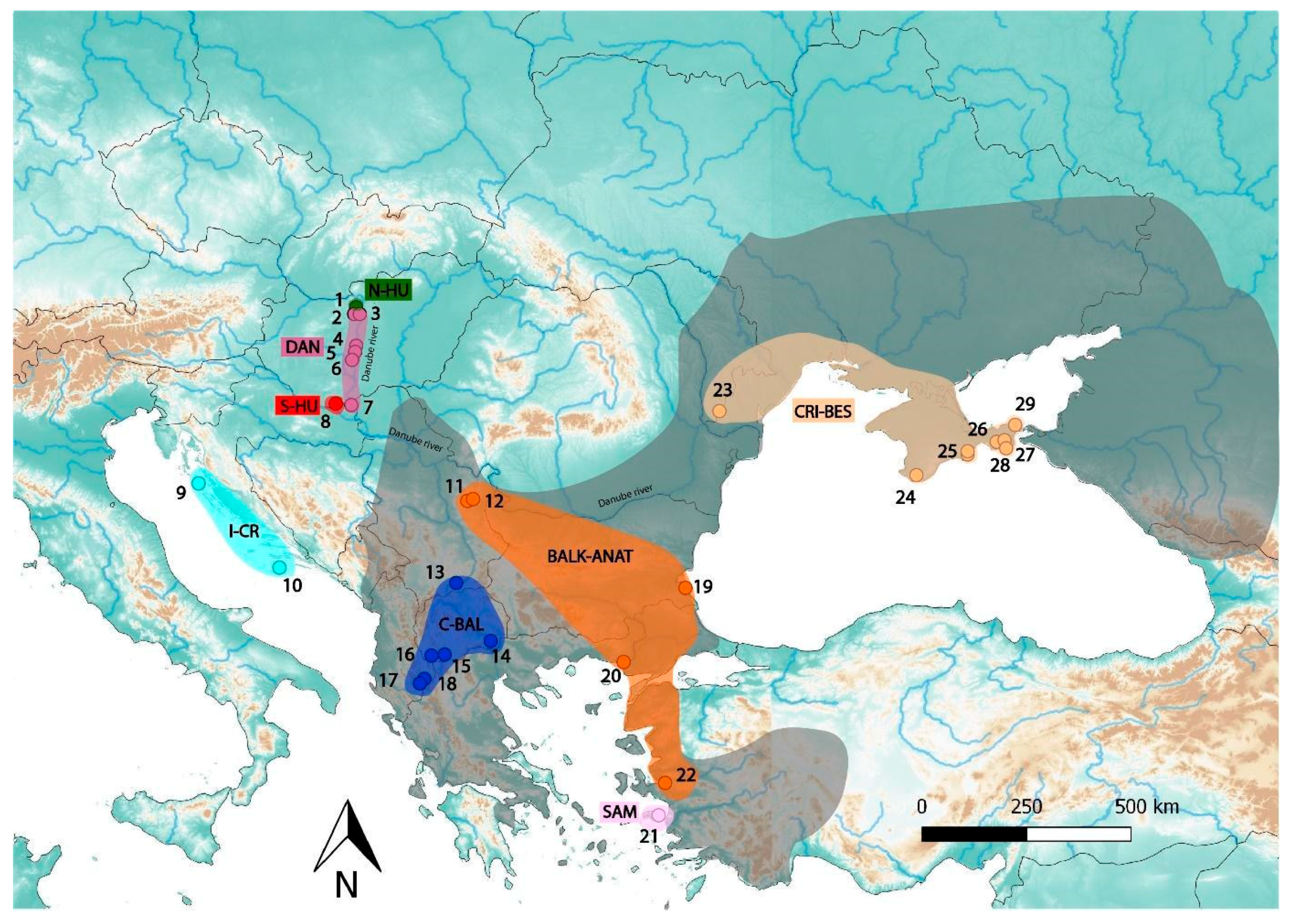
2.1. Samples and DNA Extraction
2.2. Double-Digest Restriction-Site Associated DNA (ddRAD) Sequencing and SNP Filtering
2.3. Population Summary Statistics, Structure and Differentiation
2.4. Estimating Effective Migration Surfaces (EEMS)
2.5. Landscape Genomic Analysis
2.5.1. Selection of Environmental Variables
2.5.2. Correlation Analysis of Environmental Variables
2.5.3. Samßada Analysis
3. Results
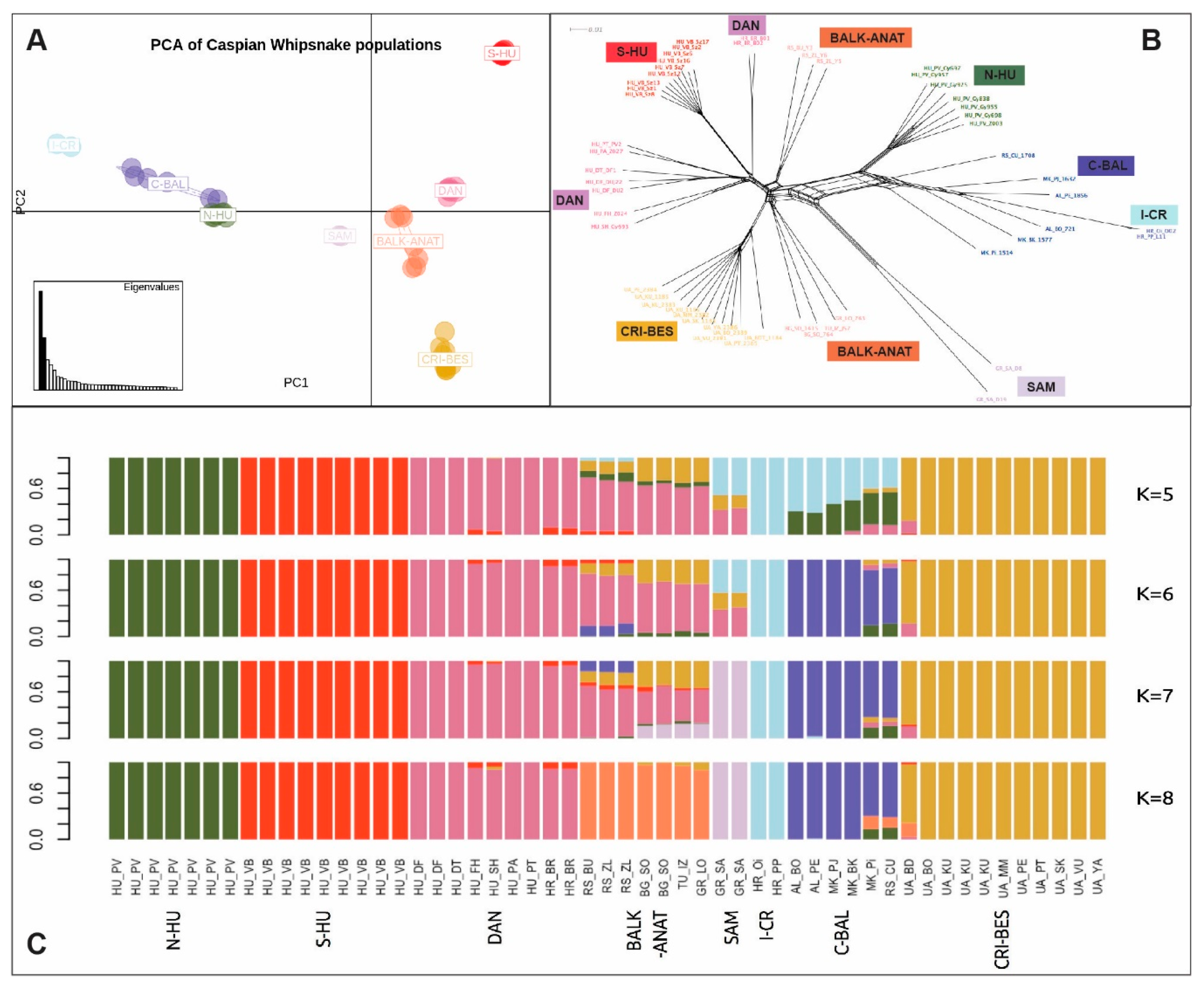
3.1. Population Structure and Genetic Differentiation among D. caspius Populations across the Entire Distribution Range
3.1.1. Overall Genetic Diversity
3.1.2. Population Structure and Admixture
3.1.3. Population Differentiation
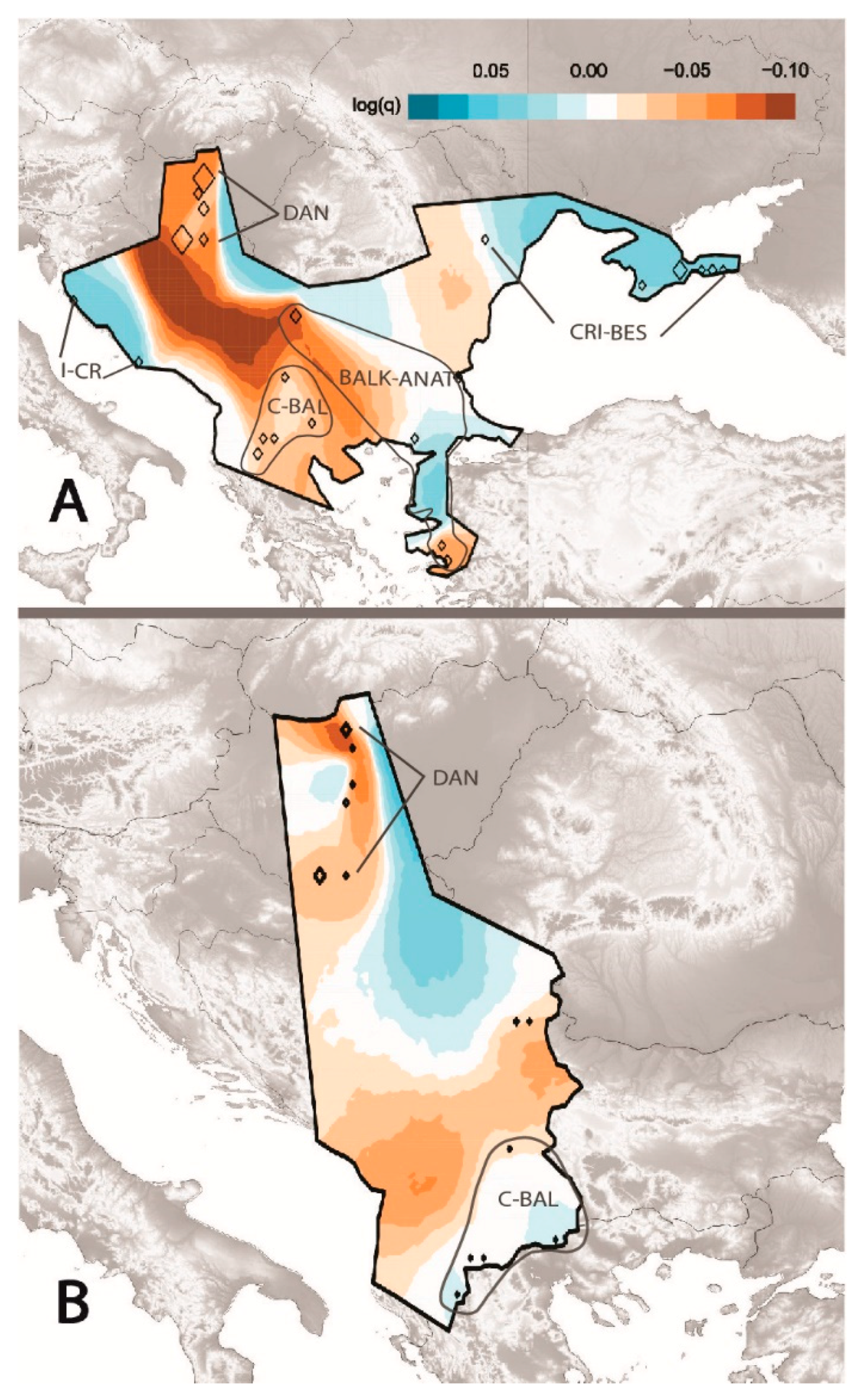
3.2. Estimated Effective Migration among D. caspius Populations
3.3. Relationship between Genotypes and Environmental Variables
4. Discussion
4.1. Structured Population and Effective Migration Rates in D. caspius across Two Continents
4.2. Environmental Adaptation in D. caspius
4.3. Conclusions and Conservation Management Recommendations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability Statement
References
- IPBES. Summary for Policymakers of the Global Assessment Report on Biodiversity and Ecosystem Services of the Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services; Díaz, S., Settele, J., Brondízio, E.S., Ngo, H.T., Guèze, M., Agard, J., Arneth, A., Balvanera, P., Brauman, K.A., Butchart, S.H.M., et al., Eds.; IPBES Secretariat: Bonn, Germany, 2019. [Google Scholar] [CrossRef]
- Barrett, R.D.; Schluter, D. Adaptation from standing genetic variation. Trends Ecol. Evol. 2008, 23, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Elmer, K.R.; Meyer, A. Adaptation in the age of ecological genomics: Insights from parallelism and convergence. Trends Ecol. Evol. 2011, 26, 298–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, D.R.; Parigi, A.; Fish, J.A.; Dworkin, I.; Wagner, A.P. The roles of standing genetic variation and evolutionary history in determining the evolvability of anti-predator strategies. PLoS ONE 2014, 9, e100163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orsini, L.; Andrew, R.; Eizaguirre, C. Evolutionary Ecological Genomics. Mol. Ecol. 2013, 22, 527–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luikart, G.; England, P.R.; Tallmon, D.; Jordan, S.; Taberlet, P. The power and promise of population genomics: From genotyping to genome typing. Nat. Rev. Genet. 2003, 4, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Joost, S.; Bonin, A.; Bruford, M.W.; Despres, L.; Conord, C.; Erhardt, G.; Taberlet, P. A spatial analysis method (SAM) to detect candidate loci for selection: Towards a landscape genomics approach to adaptation. Mol. Ecol. 2007, 16, 3955–3969. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, X.-X.; Mao, R.-L.; Yang, J.; Miao, C.-Y.; Li, Z.; Qiu, Y.-X. Ten years of landscape genomics: Challenges and opportunities. Front. Plant Sci. 2017, 8, 2136. [Google Scholar] [CrossRef] [Green Version]
- Schcherbak, N.N.; Böhme, W. Coluber caspius Gmelin, 1789—Kaspische Pfeilnatter oder Springnatter. In Handbuch der Reptilien und Amphibien Europas, Band 3/I, Schlangen (Serpentes) I; Böhme, W., Ed.; AULA-Verlag: Wiesbaden, Germany, 1993; pp. 83–96. [Google Scholar]
- Budak, A.; Göçmen, B. Herpetology, 2nd ed.; Ege Üniversitesi Basimevi: Izmir, Turkey, 2005; ISBN 975-483-658-2. [Google Scholar]
- Nagy, Z.T.; Bellaagh, M.; Wink, M.; Paunović, A.; Korsós, Z. Phylogeography of the Caspian whipsnake in Europe with emphasis on the westernmost populations. Amphib. Reptil. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Sahlean, T.C.; Strugariu, A.; Zamfirescu, S.R.; Chisamera, G.; Stanciu, C.R.; Gavril, V.D.; Gherghel, I. Filling the gaps: Updated distribution of the Caspian Whip Snake (Dolichophis caspius, Reptilia: Colubridae) in Romania. Russ. J. Herpetol. 2019, 26, 305–308. [Google Scholar] [CrossRef]
- Kletečki, E.; Lanszki, J.; Trócsányi, B.; Mužinić, J.; Purger, J.J. First record of Dolichophis caspius (Gmelin, 1789), (Reptilia: Colubridae) on the island of Olib, Croatia. Nat. Croat. 2009, 18, 437–442. [Google Scholar]
- Kreiner, G. The Snakes of Europe; Chiamira: Frankfurt am Main, Germany, 2007; ISBN 9783899734751. [Google Scholar]
- Bellaagh, M.; Korsós, Z.; Szelényi, G. New occurences of the Caspian Whipsnake, Dolichophis caspius (Reptilia: Serpentes: Colubridae) along the River Danube in Hungary. Acta Zool. Bulg. 2008, 60, 213–217. [Google Scholar]
- Covaciu-Marcov, S.; David, A. Dolichophis caspius (Serpentes: Colubridae) in Romania: New distribution records from the northern limit of its range. Turk. J. Zool. 2010, 34, 119–121. [Google Scholar] [CrossRef]
- Sahlean, T.C.; Gherghel, I.; Papeş, M.; Strugariu, A.; Zamfirescu, Ş.R. Refining climate change projections for organisms with low dispersal abilities: A case study of the Caspian whip snake. PLoS ONE 2014, 9, e91994. [Google Scholar] [CrossRef]
- Kukushkin, O.V.; Trofimov, A.G.; Turbanov, I.S.; Slodkevich, V.Y. Herpetofauna of Sevastopol city (southwestern Crimea): Species composition, zoogeographic analysis, landscape-zonal distribution, current status and protection. Ecosyst. Transform. 2019, 2, 4–62. [Google Scholar] [CrossRef]
- Babocsay, G.; Vági, B. Disappearing large whip snakes—Increasing citizen involvement in the Amphibian and Reptile Conservation Group of BirdLife Hungary. Természetvédelmi Közlemények 2012, 18, 4–44. [Google Scholar]
- Bellaagh, M.; Bakó, B. Protection Plan for the Caspian Whipsnake (Coluber caspius) in Hungary; Ministry of Environment and Water Office for Nature Conservation: Budapest, Hungary, 2004.
- Tytar, V.M.; Nekrasova, O.D. Species distribution modeling of the Caspian whipsnake Dolichophis caspius (Squamata: Serpentes): A tool for ranking conservation priorities in the Western Pontic Steppe. Biol. Commun. 2016, 3, 144–149. [Google Scholar] [CrossRef] [Green Version]
- Cox, N.A.; Temple, H.J. European Red List of Reptiles; Office for Official Publications of the European Communities: Luxembourg; Cambridge Publishers: Cambridge, UK, 2009; ISBN 978-92-79-11357-4. [Google Scholar]
- Chiucchi, J.E.; Gibbs, H.L. Similarity of contemporary and historical gene flow among highly fragmented populations of an endangered rattlesnake. Mol. Ecol. 2010, 19, 5345–5358. [Google Scholar] [CrossRef]
- Lukoschek, V. Population declines, genetic bottlenecks and potential hybridization in sea snakes on Australia’s Timor Sea reefs. Biol. Conserv. 2018, 225, 66–79. [Google Scholar] [CrossRef]
- Újvári, B.; Madsen, T.; Kotenko, T.; Olsson, M.; Shine, R.; Wittzell, H. Low genetic diversity threatens imminent extinction for the Hungarian Meadow Viper (Vipera ursinii rakosiensis). Biol. Conserv. 2002, 105, 127–130. [Google Scholar] [CrossRef]
- Vonk, F.J.; Casewell, N.R.; Henkel, C.V.; Heimberg, A.M.; Jansen, H.J.; McCleary, R.J.; Kerkkamp, H.M.E.; Vos, R.A.; Guerreiro, I.; Calvete, J.J.; et al. The king cobra genome reveals dynamic gene evolution and adaptation in the snake venom system. Proc. Natl. Acad. Sci. USA 2013, 110, 20651–20656. [Google Scholar] [CrossRef] [Green Version]
- Zinner, H. Systematics and Evolution of the Species Group Coluber jugularis Linnaeus, 1758—Coluber caspius Gmelin, 1789 (Reptilia, Serpentes). Ph.D. Thesis, Hebrew University, Jerusalem, Israel, 1972. [Google Scholar]
- Bellaagh, M.; Lazányi, I.; Korsós, Z. Calculation of fluctuating asymmetry of the biggest Caspian whipsnake population in Hungary compared to a common snake species. Biologia 2010, 65, 140–144. [Google Scholar] [CrossRef]
- Klenina, A.A. Morphology of caspian whipsnake Hierophis caspius (Gmelin, 1789) (Reptilia: Colubridae) in the Lower Volga region. Curr. Stud. Herpetol. 2015, 15, 63–68. [Google Scholar]
- Franklinos, L.H.V.; Lorch, J.M.; Bohuski, E.; Rodriguez-Ramos Fernandez, J.; Wright, O.N.; Fitzpatrick, L.; Petrovan, S.; Durrant, C.; Linton, C.; Baláž, V.; et al. Emerging fungal pathogen Ophidiomyces ophiodiicola in wild European snakes. Sci. Rep. 2017, 7, 3844. [Google Scholar] [CrossRef] [PubMed]
- Luck, G.W.; Daily, G.C.; Ehrlich, P.R. Population diversity and ecosystem services. Trends Ecol. Evol. 2003, 18, 331–336. [Google Scholar] [CrossRef] [Green Version]
- Allendorf, F.W.; Luikart, G.; Aitken, S.N. Conservation and the Genetics of Populations, 2nd ed.; Wiley-Blackwell: Oxford, UK, 2013; ISBN 978-0-470-67146-7. [Google Scholar]
- Sillero, N.; Campos, J.; Bonardi, A.; Corti, C.; Creemers, R.; Crochet, P.; Crnobrnja-Isailovic, J.; Denoël, M.; Ficetola, G.F.; Gonçalves, J.; et al. Updated distribution and biogeography of amphibians and reptiles of Europe. Amphib. Reptil. 2014, 35, 1–31. [Google Scholar] [CrossRef] [Green Version]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef] [Green Version]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daily, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Paradis, E. Pegas: An R package for population genetics with an integrated-modular approach. Bioinformatics 2010, 26, 419–420. [Google Scholar] [CrossRef] [Green Version]
- RStudio Team. RStudio: Integrated Development for R; RStudio Team: Boston, MA, USA, 2015; Available online: http://www.rstudio.com/ (accessed on 1 October 2017.).
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017; Available online: http://www.R-project.org/ (accessed on 1 October 2017.).
- Jombart, T. Adagenet: An R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Alexander, D.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Resour. 2015, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Res. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Petkova, D.; Novembre, J.; Stephens, M. Visualizing spatial population structure with estimated effective migration surfaces. Nat. Genet. 2016, 48, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Fick, S.E.; Hijmans, R.J. Worldclim 2: New 1-km spatial resolution climate surfaces for global land areas. Int. J. Climatol. 2017, 37, 4302–4315. [Google Scholar] [CrossRef]
- Prunier, J.G.; Colyn, M.; Legendre, X.; Nimons, K.F.; Flamand, C. Multicollinearity in spatial genetics: Separating the wheat from the chaff using commonality analyses. Mol. Ecol. 2015, 24, 263–283. [Google Scholar] [CrossRef]
- Belsley, D.A.; Kuh, E.; Welsch, R.E. Regression Diagnostics. Identifying Influential Data and Sources of Collinearity; John Wiley & Sons: New York, NY, USA, 1980; ISBN 9780471058564. [Google Scholar]
- Stucki, S.; Orozco-terWengel, P.; Forester, B.R.; Duruz, S.; Colli, L.; Joost, S.; Negrini, R.; Landguth, E.; Jones, M.R.; Bruford, M.W.; et al. High performance computation of landscape genomic models including local indicators of spatial association. Mol. Ecol. Res. 2017, 17, 1072–1089. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, M. fmsb: Functions for Medical Statistics Book with Some Demographic Data. R Package Version 0.7.0. 2019. Available online: https://CRAN.R-project.org/package=fmsb (accessed on 1 September 2019.).
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Fiddes, I.T.; Armstrong, J.; Diekhans, M.; Nachtweide, S.; Kronenberg, Z.N.; Underwood, J.G.; Gordon, D.; Earl, D.; Keane, T.; Eichler, E.E.; et al. Comparative Annotation Toolkit (CAT)-simultaneous clade and personal genome annotation. Genome Resour. 2018, 28, 1029–1038. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, M.S.; Ignizio, D.A. Bioclimatic Predictors for Supporting Ecological Applications in the Conterminous United States. US Geol. Surv. Data Ser. 2012, 691, 10. [Google Scholar] [CrossRef]
- Fischer, J.; Lindenmayer, D.B. Landscape modification and habitat fragmentation: A synthesis. Glob. Ecol. Biogeogr. 2007, 16, 265–280. [Google Scholar] [CrossRef]
- Heller, N.E.; Zavaleta, E.S. Biodiversity management in the face of climate change: A review of 22 years of recommendations. Biol. Conserv. 2009, 142, 14–32. [Google Scholar] [CrossRef]
- Joger, U.; Fritz, U.; Guicking, D.; Kalyabina-Hauf, S.; Nagy, Z.T.; Wink, M. Phylogeography of western Palaearctic reptiles—Spatial and temporal speciation patterns. Zool. Anz. 2007, 246, 293–313. [Google Scholar] [CrossRef]
- Hewitt, G.M. The genetic legacy of the Quaternary ice ages. Nature 2000, 405, 907–913. [Google Scholar] [CrossRef]
- Hewitt, G.M. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol. J. Linn. Soc. 1996, 58, 247–276. [Google Scholar] [CrossRef]
- Schmitt, T. Molecular biogeography of Europe: Pleistocene cycles and postglacial trends. Front. Zool. 2007, 4, e11. [Google Scholar] [CrossRef] [Green Version]
- Frank, K.; Majer, J.; Dudás, G.Y. Capture-recapture data of Large Whip Snakes Dolichophis caspius (GMELIN, 1789), in southern Transdanubia, Hungary. Herpetozoa 2012, 25, 68–71. [Google Scholar]
- Kindler, C.; Graciá, E.; Fritz, U. Extra-Mediterranean glacial refuges in barred and common grass snakes (Natrix helvetica, N. natrix). Sci. Rep. 2018, 8, 1821. [Google Scholar] [CrossRef] [Green Version]
- Werner, F. Reptilien. In Beiträge zur Kenntniss der Fauna Einiger Dalmatinischer Inseln; Galvagni, E., Ed.; Verhandlunden der Zoologisch-Botanischen Gesellschaft in Wien: Vienna, Austria, 1902; pp. 362–388. [Google Scholar]
- Musilová, R.; Zavadil, V.; Marková, S.; Kotlík, P. Relics of the Europe’s warm past: Phylogeography of the Aesculapian snake. Mol. Phylogenet. Evol. 2010, 57, 1245–1252. [Google Scholar] [CrossRef]
- Allentoft, M.E.; Rassmussen, A.R.; Kristensen, H.V. Centuries-Old DNA from an extinct population of Aesculapian snake (Zamenis longissimus) offers new phylogeographic insight. Diversity 2018, 10, 14. [Google Scholar] [CrossRef] [Green Version]
- Salvi, D.; Mendes, J.; Carranza, S.; Harris, D.J. Evolution, biogeography and systematics of the western Palearctic Zamenis ratsnakes. Zool. Scr. 2018, 47, 441–461. [Google Scholar] [CrossRef]
- Jablonski, D.; Nagy, Z.T.; Avci, A.; Olgun, K.; Kukushkin, O.V.; Safaei-Mahroo, B.; Jandzik, D. Cryptic diversity in the smooth snake (Coronella austriaca). Amphib. Reptil. 2019, 40, 179–192. [Google Scholar] [CrossRef]
- Huang, C.L.; Chen, J.H.; Chang, C.T.; Chung, J.D.; Liao, P.C.; Wang, J.C.; Hwang, S.Y. Disentangling the effects of isolation-by-distance and isolation-by-environment on genetic differentiation among Rhododendron lineages in the subgenus Tsutsusi. Tree Genet. Genomes 2016, 12, 53. [Google Scholar] [CrossRef]
- Mulin, J.L.; Bin, Y.; Pak, C.S.; Junwen, W. Exploring the function of genetic variants in the non-coding genomic regions: Approaches for identifying human regulatory variants affecting gene expression. Brief. Bioinform. 2015, 16, 393–412. [Google Scholar] [CrossRef] [Green Version]
- Aubret, F.; Shine, R. Thermal plasticity in young snakes: How will climate change affect the thermoregulatory tactics of ectotherms? J. Exp. Biol. 2010, 213, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Kearney, M.; Shine, R.; Porter, W. The potential for behavioural thermoregulation to buffer “cold-blooded” animals against climate warming. Proc. Natl. Acad. Sci. USA 2009, 106, 3835–3840. [Google Scholar] [CrossRef] [Green Version]
- Clusella-Trullas, S.; Blackburn, T.; Chown, S. Climatic predictors of temperature performance curve parameters in ectotherms imply complex responses to climate change. Am. Nat. 2011, 177, 738–751. [Google Scholar] [CrossRef] [Green Version]
- Paaijmans, K.P.; Heinig, R.L.; Seliga, R.A.; Blanford, J.I.; Blanford, S.; Murdock, C.C.; Thomas, M.B. Temperature variation makes ectotherms more sensitive to climate change. Glob. Chang. Biol. 2013, 19, 2373–2380. [Google Scholar] [CrossRef] [Green Version]
- Easterling, D.R.; Meehl, J.; Parmesan, C.; Chagnon, S.; Karl, T.R.; Mearns, L.O. Climate extremes: Observations, modeling, and impacts. Science 2000, 289, 2068–2074. [Google Scholar] [CrossRef] [Green Version]
- Gaudet, P.; Livstone, M.S.; Lewis, S.E.; Thomas, P.D. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Brief. Bioinform. 2011, 12, 449–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Population Name | Locality ID on Figure 1 | Sample ID | Sample Origin | Locality | Country | Lat | Long |
|---|---|---|---|---|---|---|---|
| N-HU | 1 | HU_BU_HU_Gy697 | Tissue (muscle) | Budapest, Hűvösvölgy | Hungary | 47.5399 | 18.9661 |
| 1 | HU_BU_PV_Gy698 | Tissue (muscle) | Budapest, Vöröskővár | Hungary | 47.5561 | 18.9768 | |
| 1 | HU_BU_PV_Gy838 | Buccal swab | Budapest, Vöröskővár | Hungary | 47.5561 | 18.9768 | |
| 1 | HU_BU_PV_Gy925 | Buccal swab | Budapest, Vöröskővár | Hungary | 47.5562 | 18.9763 | |
| 1 | HU_BU_PV_Gy955 | Shed skin | Budapest, Vöröskővár | Hungary | 47.5558 | 18.9767 | |
| 1 | HU_BU_PV_Gy957 | Buccal swab | Budapest, Vöröskővár | Hungary | 47.5561 | 18.9768 | |
| 1 | HU_BU_PV_Z003 | Shed skin | Budapest, Vöröskővár | Hungary | 47.5557 | 18.9752 | |
| DAN | 2 | HU_BU_SH_Gy693 | Tissue (liver) | Budapest, Sas Hill | Hungary | 47.4821 | 19.0196 |
| 3 | HU_BU_FH_Z024 | Shed skin | Budapest, Farkas Hill | Hungary | 47.4724 | 18.9427 | |
| 4 | HU_DF_DU2 | Blood | Dunaújváros | Hungary | 46.9106 | 18.9461 | |
| 4 | HU_DF_DUJ22 | Blood | Dunaújváros | Hungary | 46.9106 | 18.9461 | |
| 5 | HU_DT_DF1 | Blood | Dunaföldvár | Hungary | 46.8027 | 18.9406 | |
| 6 | HU_PT_PV2 | Blood | Paks | Hungary | 46.6626 | 18.8605 | |
| 6 | HU_TO_PA_Z027 | Shed skin | Paks | Hungary | 46.6626 | 18.8605 | |
| 8 | HR_BR_B01 | Shed skin | Batina | Croatia | 45.8334 | 18.8382 | |
| 8 | HR_BR_B02 | Shed skin | Batina | Croatia | 45.8334 | 18.8387 | |
| S-HU | 7 | HU_VB_Sz1 | Blood | Villány | Hungary | 45.8571 | 18.4185 |
| 7 | HU_VB_Sz12 | Blood | Villány | Hungary | 45.8571 | 18.4185 | |
| 7 | HU_VB_Sz13 | Blood | Villány | Hungary | 45.8571 | 18.4185 | |
| 7 | HU_VB_Sz16 | Blood | Villány | Hungary | 45.8571 | 18.4185 | |
| 7 | HU_VB_Sz17 | Blood | Villány | Hungary | 45.8571 | 18.4185 | |
| 7 | HU_VB_Sz2 | Blood | Villány | Hungary | 45.8571 | 18.4185 | |
| 7 | HU_VB_Sz6 | Blood | Villány | Hungary | 45.8571 | 18.4185 | |
| 7 | HU_VB_Sz7 | Blood | Villány | Hungary | 45.8571 | 18.4185 | |
| 7 | HU_VB_Sz8 | Blood | Villány | Hungary | 45.8571 | 18.4185 | |
| I-CR | 9 | HR_LA_Oi_O02 | Blood | Olib island | Croatia | 44.3656 | 14.7855 |
| 10 | HR_LA_PP_L11 | Blood | Lastovo island | Croatia | 42.7532 | 16.9169 | |
| BALK-ANAT | 11 | RS_ZL_Y5 | Blood | Zlot | Serbia | 44.0387 | 21.9300 |
| 11 | RS_ZL_Y6 | Blood | Zlot | Serbia | 44.0387 | 21.9300 | |
| 12 | RS_BU_Y3 | Blood | Brestovacka | Serbia | 44.0621 | 22.0497 | |
| 19 | BG_SO_1415 | Shed skin | Sozopol | Bulgaria | 42.3955 | 27.6996 | |
| 19 | BG_SO_764 | Tissue (muscle) | near Sozopol | Bulgaria | 42.4104 | 27.6497 | |
| 20 | GR_LO_763 | Tissue (muscle) | Loutros | Greece | 40.8806 | 26.0458 | |
| 22 | TU_IZ_J57 | Tissue (muscle) | Izmir | Turkey | 38.4237 | 27.1428 | |
| C-BAL | 13 | RS_CU_1708 | Blood | Cukarka | Serbia | 42.2874 | 21.7082 |
| 14 | MK_Pi_1514 | Tissue (muscle) | Pirava | North Macedonia | 41.3080 | 22.5356 | |
| 15 | MK_BK_1577 | Tissue (muscle) | Bilbil Kamen | North Macedonia | 41.0398 | 21.2997 | |
| 16 | MK_PPj_1632 | Tissue (muscle) | Pokrvenik, Prespansko jezero | North Macedonia | 41.0150 | 20.9648 | |
| 17 | AL_BO_721 | Tissue (muscle) | Boboshticë | Albania | 40.5505 | 20.7597 | |
| 18 | AL_PE_1856 | Tissue (muscle) | Pepellash | Albania | 40.4619 | 20.6672 | |
| SAM | 21 | GR_SA_D19 | Tissue (muscle) | Samos | Greece | 37.7547 | 26.9777 |
| 21 | GR_SA_D8 | Tissue (muscle) | Samos | Greece | 37.7547 | 26.9777 | |
| CRI-BES | 23 | UA_BDT_1184 | Tissue (scale) | Tabaky | Ukraine | 45.7332 | 28.6020 |
| 24 | UA_PE_2384 | Tissue (muscle) | Peredovoe | Ukraine /Crimea | 44.5339 | 33.8254 | |
| 25 | UA_MM_2382 | Tissue (muscle) | Karadag Mt. | Ukraine /Crimea | 44.9319 | 35.2212 | |
| 25 | UA_KU_1185 | Tissue (muscle) | Kurortnoe | Ukraine /Crimea | 44.9181 | 35.2028 | |
| 25 | UA_KU_1186 | Tissue (muscle) | Kurortnoe | Ukraine /Crimea | 44.9126 | 35.2006 | |
| 25 | UA_KU_2383 | Tissue (muscle) | Kurortnoe | Ukraine /Crimea | 44.9103 | 35.1625 | |
| 25 | UA_SK_1183 | Tissue (muscle) | Schebetovka | Ukraine /Crimea | 44.9496 | 35.1873 | |
| 26 | UA_VU_2391 | Tissue (scale) | Vulkanovka | Ukraine /Crimea | 45.1503 | 35.9309 | |
| 27 | UA_PT_2385 | Tissue (scale) | Ptashkino | Ukraine /Crimea | 45.1716 | 36.1635 | |
| 28 | UA_YA_2386 | Tissue (scale) | Yakovenkovo | Ukraine /Crimea | 45.0451 | 36.2412 | |
| 29 | UA_BO_2389 | Tissue (scale) | Bondarenkovo | Ukraine /Crimea | 45.4467 | 36.4346 |
| Population | DAN | S-HU | N-HU | C-BAL | BALK-ANAT | CRI-BES |
|---|---|---|---|---|---|---|
| DAN | - | |||||
| S-HU | 0.340 * | - | ||||
| N-HU | 0.420 * | 0.595 * | - | |||
| C-BAL | 0.401 * | 0.565 * | 0.261 * | - | ||
| BALK-ANAT | 0.132 * | 0.344 * | 0.321 * | 0.302 * | - | |
| CRI-BES | 0.295 * | 0.487 * | 0.465 * | 0.458 * | 0.194 * | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahtani-Williams, S.; Fulton, W.; Desvars-Larrive, A.; Lado, S.; Elbers, J.P.; Halpern, B.; Herczeg, D.; Babocsay, G.; Lauš, B.; Nagy, Z.T.; et al. Landscape Genomics of a Widely Distributed Snake, Dolichophis caspius (Gmelin, 1789) across Eastern Europe and Western Asia. Genes 2020, 11, 1218. https://doi.org/10.3390/genes11101218
Mahtani-Williams S, Fulton W, Desvars-Larrive A, Lado S, Elbers JP, Halpern B, Herczeg D, Babocsay G, Lauš B, Nagy ZT, et al. Landscape Genomics of a Widely Distributed Snake, Dolichophis caspius (Gmelin, 1789) across Eastern Europe and Western Asia. Genes. 2020; 11(10):1218. https://doi.org/10.3390/genes11101218
Chicago/Turabian StyleMahtani-Williams, Sarita, William Fulton, Amelie Desvars-Larrive, Sara Lado, Jean Pierre Elbers, Bálint Halpern, Dávid Herczeg, Gergely Babocsay, Boris Lauš, Zoltán Tamás Nagy, and et al. 2020. "Landscape Genomics of a Widely Distributed Snake, Dolichophis caspius (Gmelin, 1789) across Eastern Europe and Western Asia" Genes 11, no. 10: 1218. https://doi.org/10.3390/genes11101218
APA StyleMahtani-Williams, S., Fulton, W., Desvars-Larrive, A., Lado, S., Elbers, J. P., Halpern, B., Herczeg, D., Babocsay, G., Lauš, B., Nagy, Z. T., Jablonski, D., Kukushkin, O., Orozco-terWengel, P., Vörös, J., & Burger, P. A. (2020). Landscape Genomics of a Widely Distributed Snake, Dolichophis caspius (Gmelin, 1789) across Eastern Europe and Western Asia. Genes, 11(10), 1218. https://doi.org/10.3390/genes11101218