Genome-Wide Association Study of Root System Development at Seedling Stage in Rice


 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Whole Genotype Collection
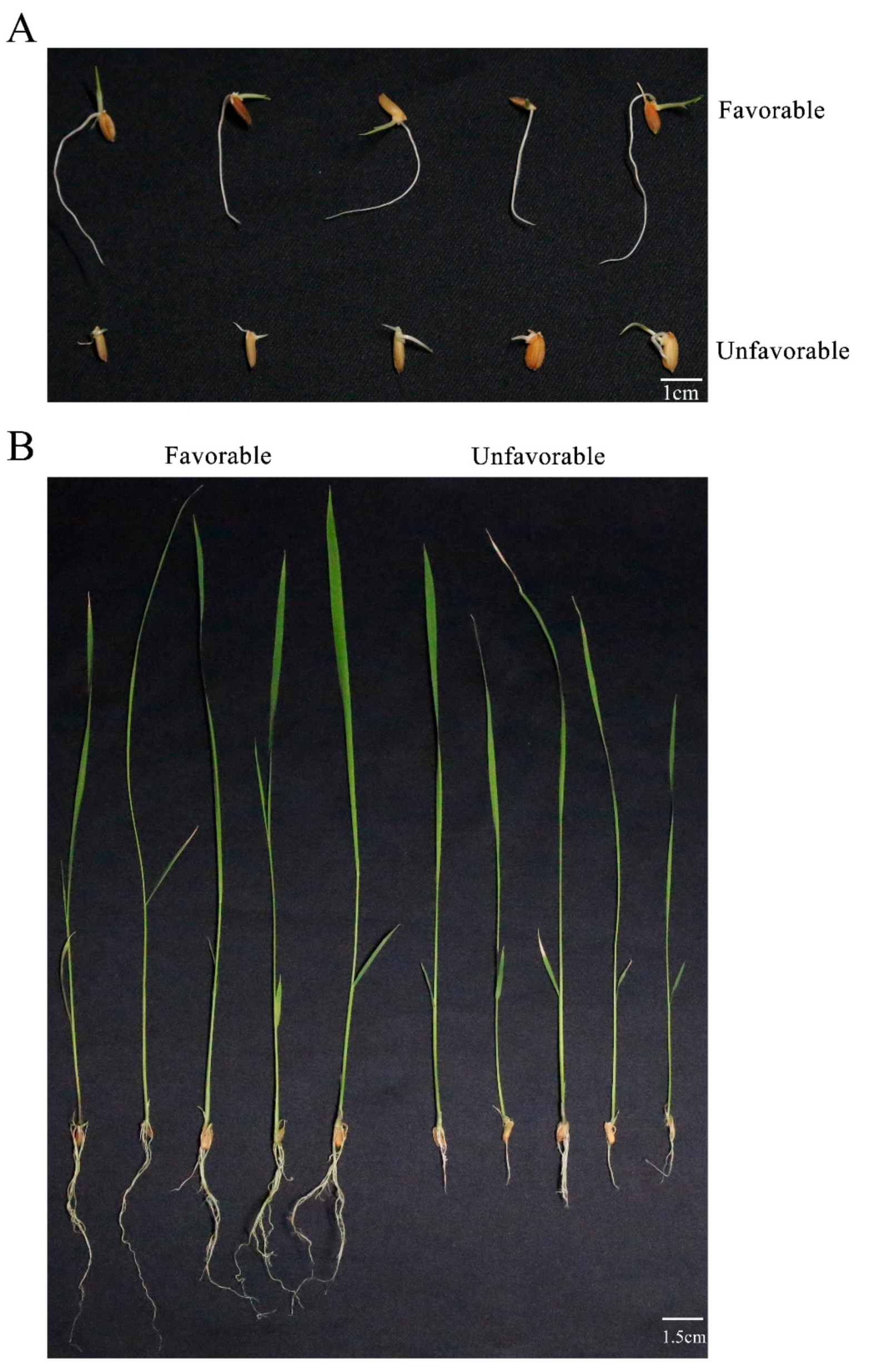
2.2. Evaluation of Root System Development
2.3. Population and Genotype Analysis
2.4. GWAS Analysis
2.5. Bioinformatics Analysis and Candidate Gene Prediction
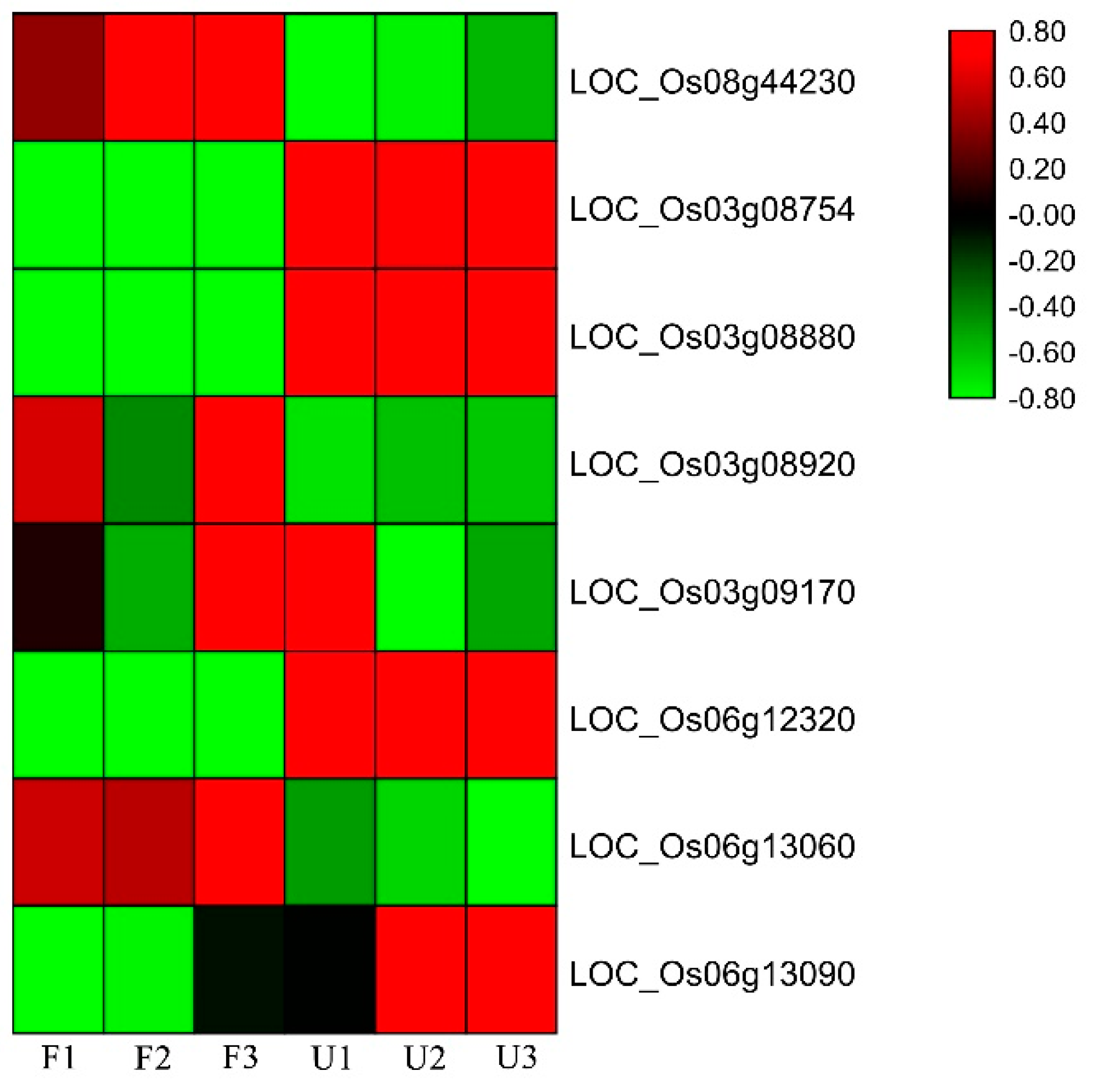
2.6. RNA Extraction, cDNA Synthesis, and Expression Analysis
2.7. Sequence and Haplotype Analysis
3. Results
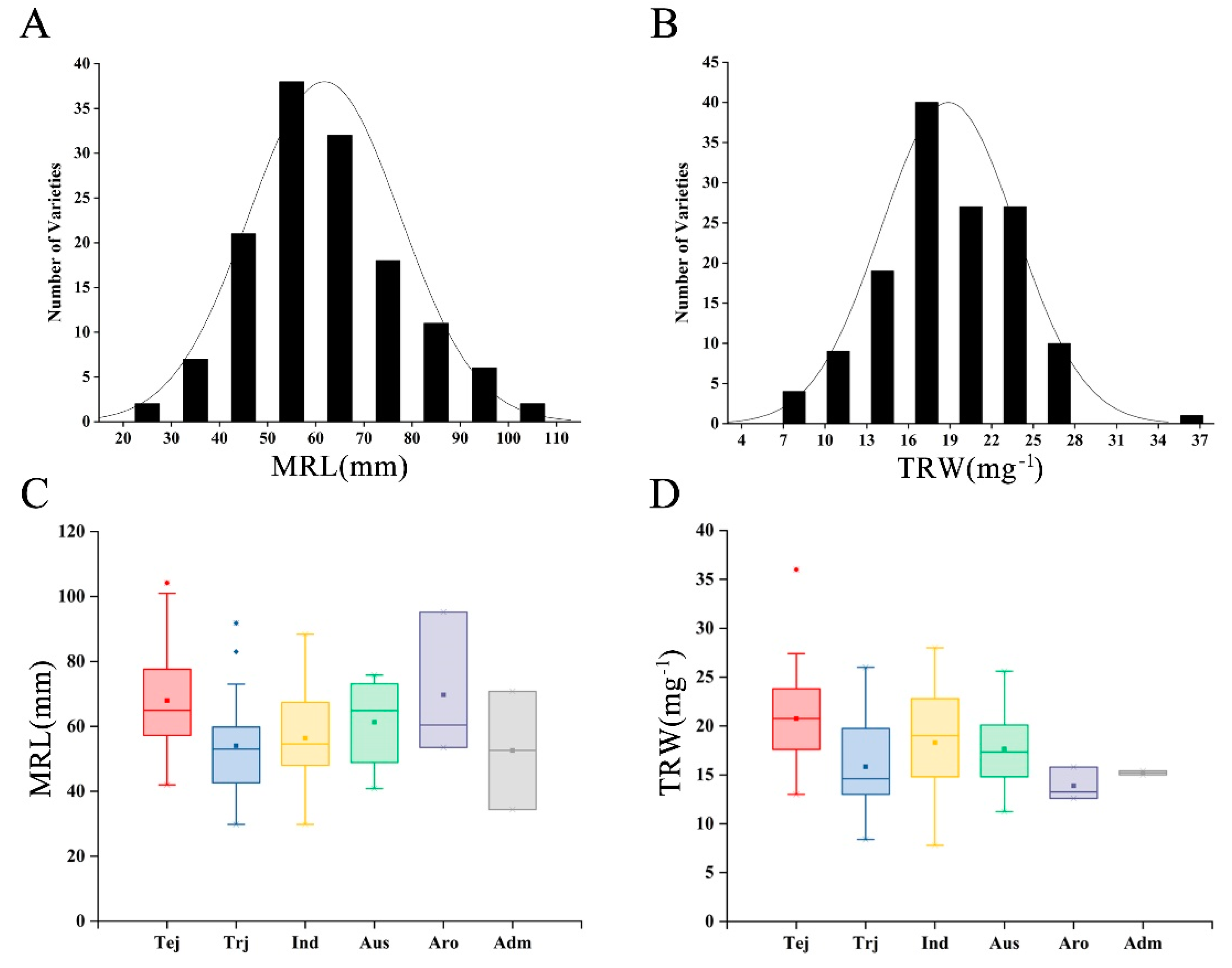
3.1. Phenotype Evaluation of Root System Development
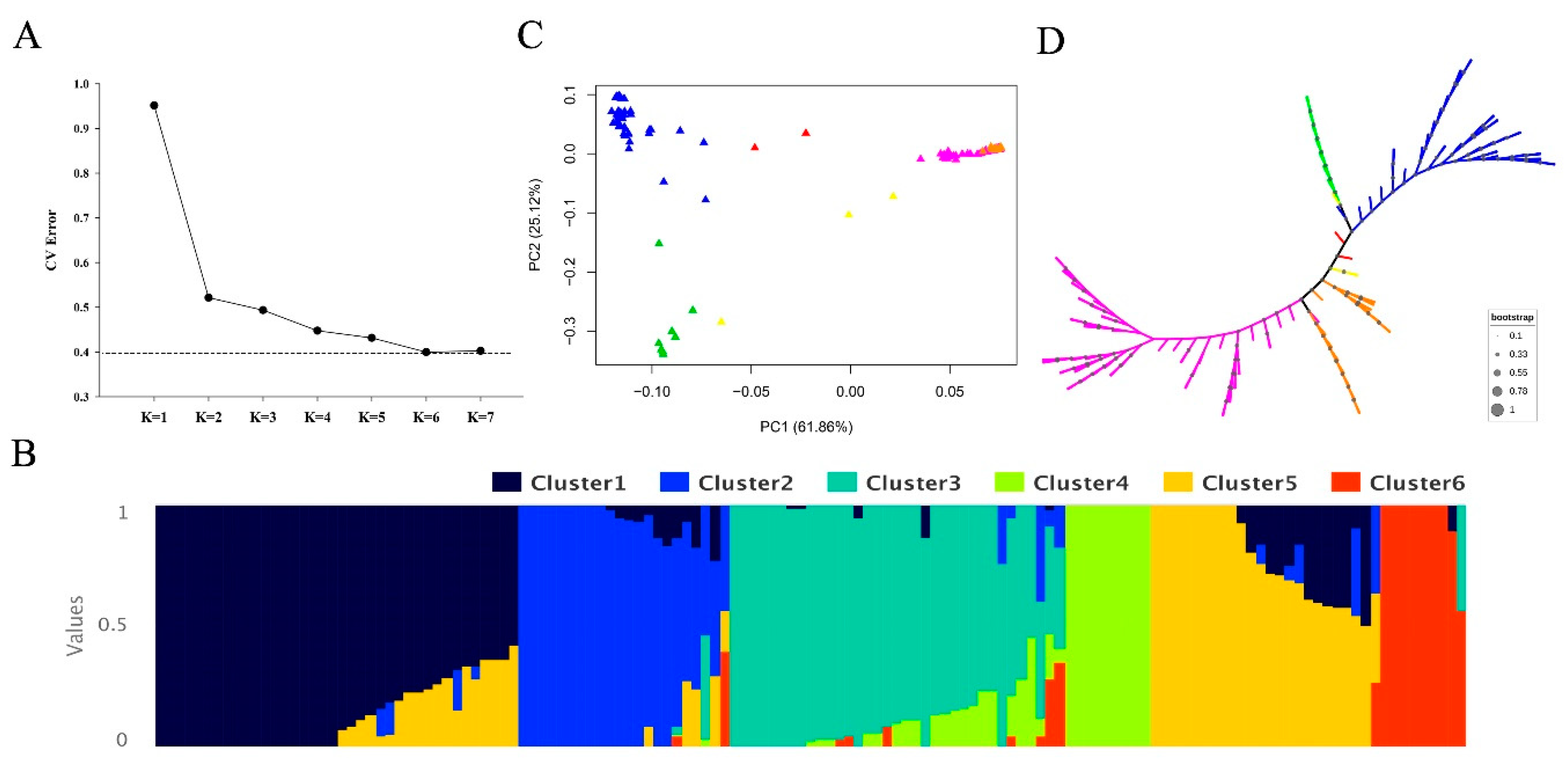
3.2. Population Structure and LD Decay Analysis
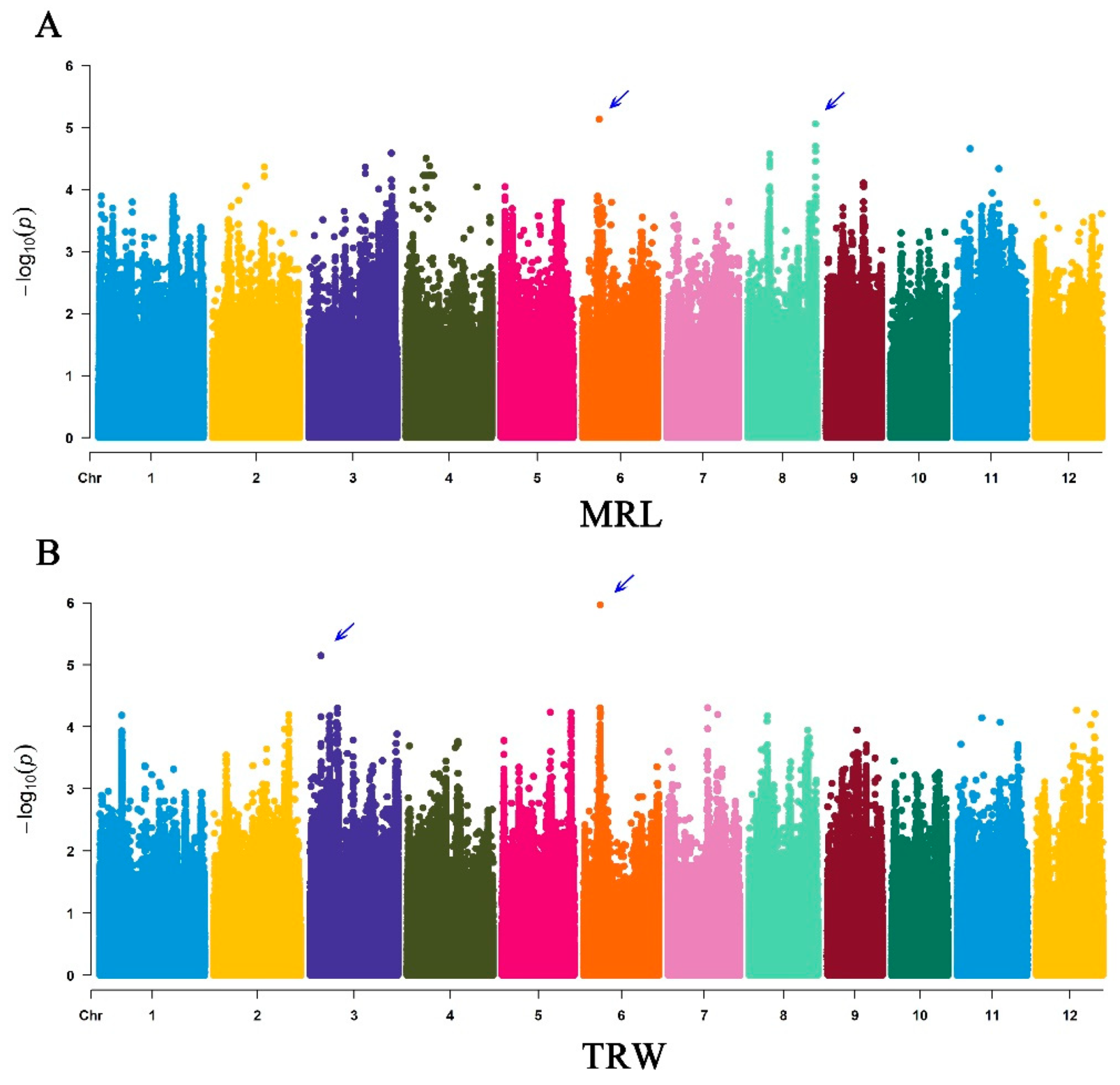
3.3. GWAS Analysis
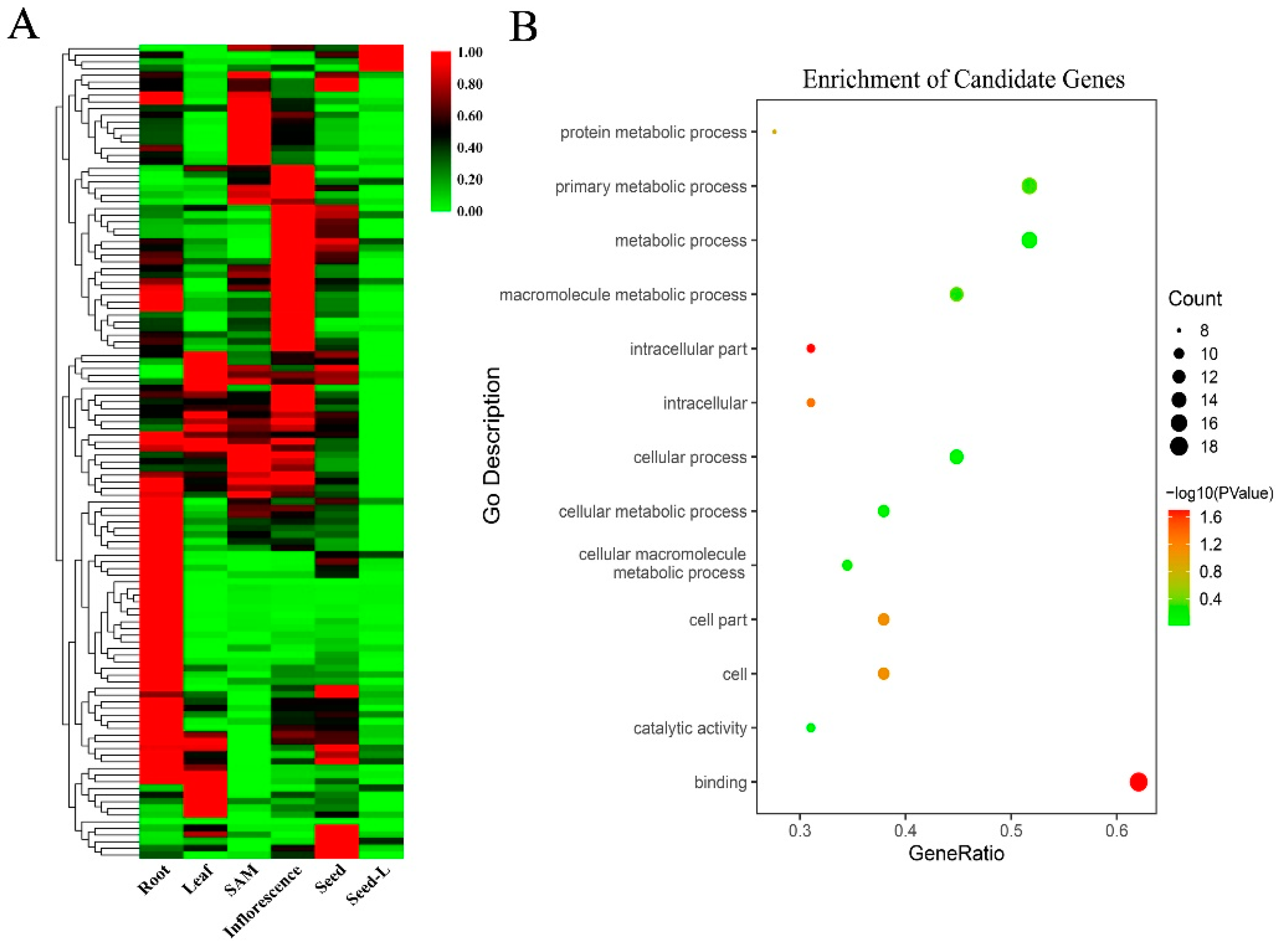
3.4. Candidate Genes and Bioinformatics Analysis
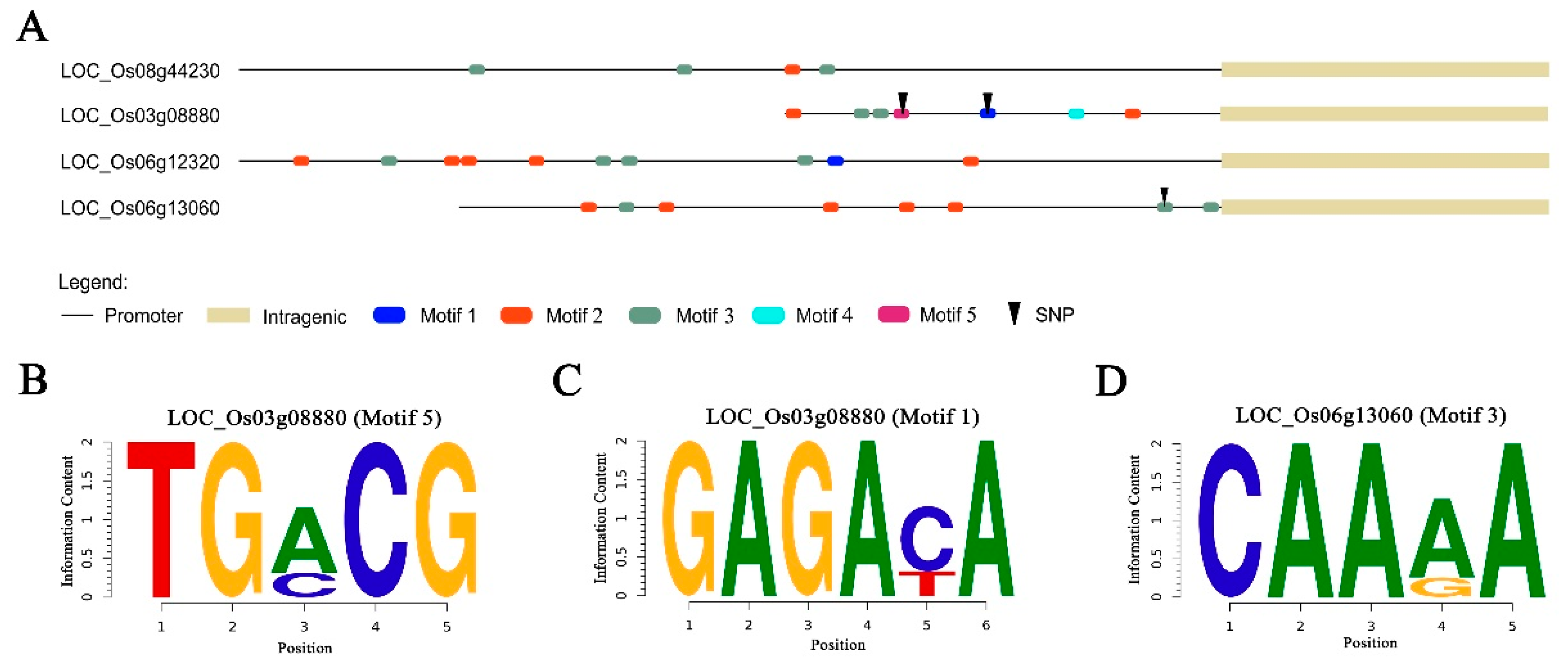
3.5. Analysis of SNP Function in Promoter Region
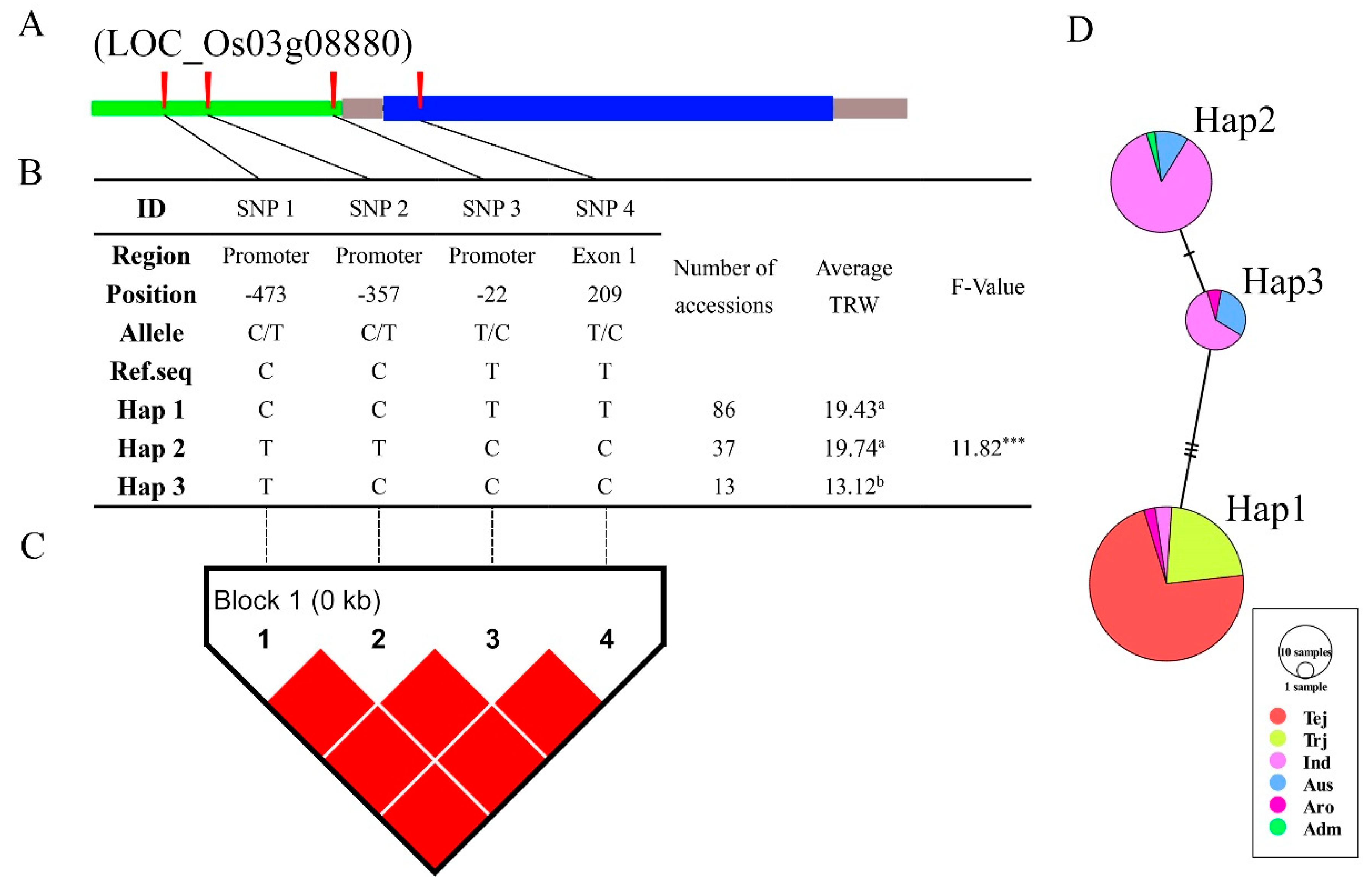
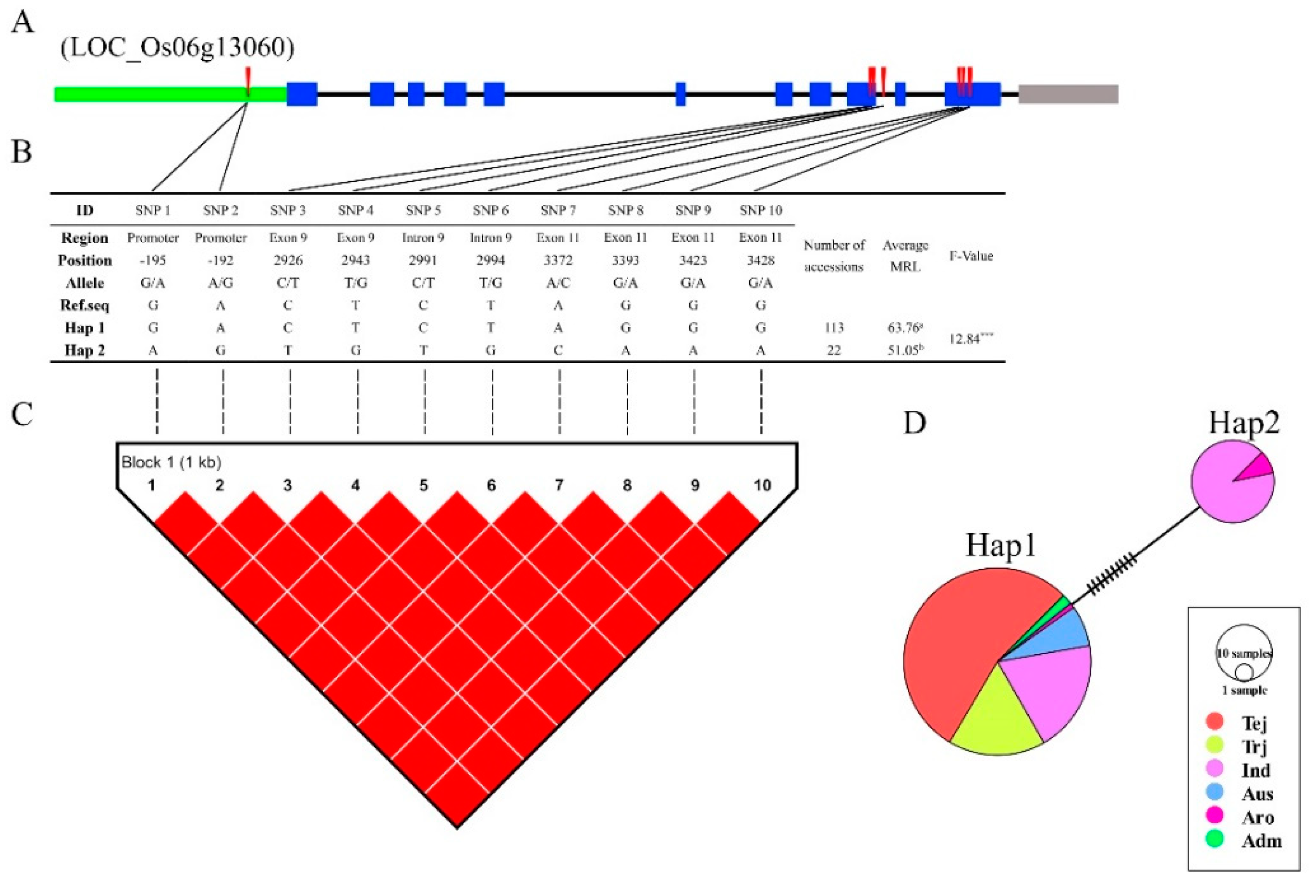
3.6. Haplotype Analysis of Reported and Novel Candidate Genes
4. Discussion
4.1. Root System Development as the Crucial Trait for Rice
4.2. Candidate Genes Identified by GWAS
4.3. LD, Haplotype, and Functional SNP Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, Q.; Wing, R. Genome studies and molecular genetics: Understanding the functional genome based on the rice model. Curr. Opin. Plant Biol. 2013, 16, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Muthayya, S.; Sugimoto, J.D.; Montgomery, S.; Maberly, G.F. An overview of global rice production, supply, trade, and consumption. Ann. N. Y. Acad. Sci. 2014, 1324, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasulu, N.; Butardo, V.M., Jr.; Misra, G.; Cuevas, R.P.; Anacleto, R.; Kavi Kishor, P.B. Designing climate-resilient rice with ideal grain quality suited for high-temperature stress. J. Exp. Bot. 2015, 66, 1737–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Dai, X.; Xu, Y.; Luo, W.; Zheng, X.; Zeng, D.; Pan, Y.; Lin, X.; Liu, H.; Zhang, D. COLD1 confers chilling tolerance in rice. Cell 2015, 160, 1209–1221. [Google Scholar] [CrossRef] [Green Version]
- Ghomi, K.; Rabiei, B.; Sabouri, H.; Sabouri, A. Mapping QTLs for traits related to salinity tolerance at seedling stage of rice (Oryza sativa L.): An agrigenomics study of an Iranian rice population. OMICS 2013, 17, 242–251. [Google Scholar] [CrossRef]
- Quero, G.; Gutiérrez, L.; Monteverde, E.; Blanco, P.; Pérez de Vida, F.; Rosas, J.; Fernández, S.; Garaycochea, S.; McCouch, S.; Berberian, N. Genome-Wide Association Study Using Historical Breeding Populations Discovers Genomic Regions Involved in High-Quality Rice. Plant Genome 2018, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Liu, D.; Zhang, G.; Gao, S.; Liu, L.; Xu, F.; Che, R.; Wang, Y.; Tong, H.; Chu, C. Big Grain3, encoding a purine permease, regulates grain size via modulating cytokinin transport in rice. J. Integr. Plant Biol. 2019, 61, 581–597. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Zhou, S.; Zhang, Q.; Song, H.; Zhou, D.-X.; Zhao, Y. Transcriptional regulatory network of WOX11 is involved in the control of crown root development, cytokinin signals, and redox in rice. J. Exp. Bot. 2017, 68, 2787–2798. [Google Scholar] [CrossRef]
- Li, X.; Guo, Z.; Lv, Y.; Cen, X.; Ding, X.; Wu, H.; Li, X.; Huang, J.; Xiong, L. Genetic control of the root system in rice under normal and drought stress conditions by genome-wide association study. PLoS Genet. 2017, 13, e1006889. [Google Scholar] [CrossRef] [Green Version]
- Okushima, Y.; Overvoorde, P.J.; Arima, K.; Alonso, J.M.; Chan, A.; Chang, C.; Ecker, J.R.; Hughes, B.; Lui, A.; Nguyen, D. Functional genomic analysis of the AUXIN RESPONSE FACTOR gene family members in Arabidopsis thaliana: Unique and overlapping functions of ARF7 and ARF19. Plant Cell 2005, 17, 444–463. [Google Scholar] [CrossRef] [Green Version]
- Coudert, Y.; Le, V.A.T.; Adam, H.; Bès, M.; Vignols, F.; Jouannic, S.; Guiderdoni, E.; Gantet, P. Identification of CROWN ROOTLESS 1-regulated genes in rice reveals specific and conserved elements of postembryonic root formation. New Phytol. 2015, 206, 243–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhu, S.; Song, X.; Shen, Y.; Chen, H.; Yu, J.; Yi, K.; Liu, Y.; Karplus, V.J.; Wu, P. A rice glutamate receptor–like gene is critical for the division and survival of individual cells in the root apical meristem. Plant Cell 2006, 18, 340–349. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Parida, S.K.; Agarwal, P.; Tyagi, A.K. Transcription factor OsNF-YB9 regulates reproductive growth and development in rice. Planta 2019, 250, 1849–1865. [Google Scholar] [CrossRef] [PubMed]
- Kikui, S.; Sasaki, T.; Maekawa, M.; Miyao, A.; Hirochika, H.; Matsumoto, H.; Yamamoto, Y. Physiological and genetic analyses of aluminium tolerance in rice, focusing on root growth during germination. J. Inorg. Biochem. 2005, 99, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Z.; Li, L.; Zhou, Q.; Xiao, Y.; Wei, X.; Zhou, M. Short-term complete submergence of rice at the tillering stage increases yield. PLoS ONE 2015, 10, e0127982. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Chen, J.; Cao, F.; Jiang, L.; Zou, Y. Root morphology was improved in a late-stage vigor super rice cultivar. PLoS ONE 2015, 10, e0142977. [Google Scholar] [CrossRef]
- Sazuka, T.; Kamiya, N.; Nishimura, T.; Ohmae, K.; Sato, Y.; Imamura, K.; Nagato, Y.; Koshiba, T.; Nagamura, Y.; Ashikari, M. A rice tryptophan deficient dwarf mutant, tdd1, contains a reduced level of indole acetic acid and develops abnormal flowers and organless embryos. Plant J. 2009, 60, 227–241. [Google Scholar] [CrossRef]
- Behringer, C.; Schwechheimer, C. B-GATA transcription factors–insights into their structure, regulation, and role in plant development. Front. Plant Sci. 2015, 6, 90. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Ma, T.; Wang, X.; Deng, Y.; Ma, H.; Zhang, R.; Zhao, J. OsAUX 1 controls lateral root initiation in rice (Oryza sativa L.). Plant Cell Environ. 2015, 38, 2208–2222. [Google Scholar] [CrossRef]
- Müller, B.; Sheen, J. Cytokinin and auxin interaction in root stem-cell specification during early embryogenesis. Nature 2008, 453, 1094–1097. [Google Scholar] [CrossRef] [Green Version]
- Inukai, Y.; Sakamoto, T.; Ueguchi-Tanaka, M.; Shibata, Y.; Gomi, K.; Umemura, I.; Hasegawa, Y.; Ashikari, M.; Kitano, H.; Matsuoka, M. Crown rootless1, which is essential for crown root formation in rice, is a target of an AUXIN RESPONSE FACTOR in auxin signaling. Plant Cell 2005, 17, 1387–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Wang, S.; Yu, X.; Yu, J.; He, X.; Zhang, S.; Shou, H.; Wu, P. ARL1, a LOB-domain protein required for adventitious root formation in rice. Plant J. 2005, 43, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhu, L.; Shou, H.; Wu, P. A PIN1 family gene, OsPIN1, involved in auxin-dependent adventitious root emergence and tillering in rice. Plant Cell Physiol. 2005, 46, 1674–1681. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Tao, J.; Bi, Y.; Hou, M.; Lou, J.; Chen, X.; Zhang, X.; Luo, L.; Xie, X.; Yoneyama, K. OsPIN1b is involved in rice seminal root elongation by regulating root apical meristem activity in response to low nitrogen and phosphate. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, J.; Wu, L.; Shao, Y.; Wu, Y.; Mao, C. Functional divergence of PIN1 paralogous genes in rice. Plant Cell Physiol. 2019, 60, 2720–2732. [Google Scholar] [CrossRef]
- Kitomi, Y.; Ogawa, A.; Kitano, H.; Inukai, Y. CRL4 regulates crown root formation through auxin transport in rice. Plant Root 2008, 2, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-F.; He, F.-F.; Ma, X.-X.; Mao, C.-Z.; Hodgman, C.; Lu, C.-G.; Wu, P. OsCAND1 is required for crown root emergence in rice. Mol. Plant 2011, 4, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Bian, H.; Xie, Y.; Guo, F.; Han, N.; Ma, S.; Zeng, Z.; Wang, J.; Yang, Y.; Zhu, M. Distinctive expression patterns and roles of the miRNA393/TIR1 homolog module in regulating flag leaf inclination and primary and crown root growth in rice (Oryza sativa). New Phytol. 2012, 196, 149–161. [Google Scholar] [CrossRef]
- Xia, K.; Wang, R.; Ou, X.; Fang, Z.; Tian, C.; Duan, J.; Wang, Y.; Zhang, M. OsTIR1 and OsAFB2 downregulation via OsmiR393 overexpression leads to more tillers, early flowering and less tolerance to salt and drought in rice. PLoS ONE 2012, 7, e30039. [Google Scholar] [CrossRef]
- Debi, B.R.; Taketa, S.; Ichii, M. Cytoinin inhibits lateral root initiation but stimulates lateral root elongation in rice (Oryza sativa). J. Plant Physiol. 2005, 162, 507–515. [Google Scholar] [CrossRef]
- Cheng, X.; Jiang, H.; Zhang, J.; Qian, Y.; Zhu, S.; Cheng, B. Overexpression of type-A rice response regulators, OsRR3 and OsRR5, results in lower sensitivity to cytokinins. Genet. Mol. Res. 2010, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Kitomi, Y.; Ito, H.; Hobo, T.; Aya, K.; Kitano, H.; Inukai, Y. The auxin responsive AP2/ERF transcription factor CROWN ROOTLESS5 is involved in crown root initiation in rice through the induction of OsRR1, a type-A response regulator of cytokinin signaling. Plant J. 2011, 67, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hu, Y.; Dai, M.; Huang, L.; Zhou, D.-X. The WUSCHEL-related homeobox gene WOX11 is required to activate shoot-borne crown root development in rice. Plant Cell 2009, 21, 736–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Chen, D.; Ren, Y.; Zhang, X.; Zhao, J. Characteristic and expression analysis of a metallothionein gene, OsMT2b, down-regulated by cytokinin suggests functions in root development and seed embryo germination of rice. Plant Physiol. 2008, 146, 1637–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Cheng, S.; Song, Y.; Huang, Y.; Zhou, S.; Liu, X.; Zhou, D.-X. The interaction between rice ERF3 and WOX11 promotes crown root development by regulating gene expression involved in cytokinin signaling. Plant Cell 2015, 27, 2469–2483. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Feng, F.; Zhang, Y.; Elesawi, I.E.; Xu, K.; Li, T.; Mei, H.; Liu, H.; Gao, N.; Chen, C. A novel rice grain size gene OsSNB was identified by genome-wide association study in natural population. PLoS Genet. 2019, 15, e1008191. [Google Scholar] [CrossRef]
- Petersdorf, E.W.; O’hUigin, C. The MHC in the era of next-generation sequencing: Implications for bridging structure with function. Hum. Immunol. 2019, 80, 67–78. [Google Scholar] [CrossRef]
- Yano, K.; Morinaka, Y.; Wang, F.; Huang, P.; Takehara, S.; Hirai, T.; Ito, A.; Koketsu, E.; Kawamura, M.; Kotake, K. GWAS with principal component analysis identifies a gene comprehensively controlling rice architecture. Proc. Natl. Acad. Sci. USA 2019, 116, 21262–21267. [Google Scholar] [CrossRef]
- Yano, K.; Yamamoto, E.; Aya, K.; Takeuchi, H.; Lo, P.-c.; Hu, L.; Yamasaki, M.; Yoshida, S.; Kitano, H.; Hirano, K. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 2016, 48, 927. [Google Scholar] [CrossRef]
- Kim, T.-S.; He, Q.; Kim, K.-W.; Yoon, M.-Y.; Ra, W.-H.; Li, F.P.; Tong, W.; Yu, J.; Oo, W.H.; Choi, B. Genome-wide resequencing of KRICE_CORE reveals their potential for future breeding, as well as functional and evolutionary studies in the post-genomic era. BMC Genom. 2016, 17, 408. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, S. Routine Procedure for Growing Rice Plants in Culture Solution; Laboratory Manual for Physiological Studies of Rice, International Rice Research Institute: Los Baños, Philippine, 1976; pp. 61–66. [Google Scholar]
- Wang, F.; Longkumer, T.; Catausan, S.C.; Calumpang, C.L.F.; Tarun, J.A.; Cattin-Ortola, J.; Ishizaki, T.; Pariasca Tanaka, J.; Rose, T.; Wissuwa, M. Genome-wide association and gene validation studies for early root vigour to improve direct seeding of rice. Plant Cell Environ. 2018, 41, 2731–2743. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, R.M. pophelper: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life v2: Online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 2011, 39, W475–W478. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, H.; Xu, J.; Jiang, C.; Yin, Z.; Xiong, H.; Xie, J.; Wang, X.; Zhu, X.; Li, Y.; et al. Loci and natural alleles underlying robust roots and adaptive domestication of upland ecotype rice in aerobic conditions. PLoS Genet. 2018, 14, e1007521. [Google Scholar] [CrossRef]
- Yang, W.; Guo, Z.; Huang, C.; Duan, L.; Chen, G.; Jiang, N.; Fang, W.; Feng, H.; Xie, W.; Lian, X.; et al. Combining high-throughput phenotyping and genome-wide association studies to reveal natural genetic variation in rice. Nat. Commun. 2014, 5, 5087. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Guo, A.-Y.; Zhu, Q.-H.; Chen, X.; Luo, J.-C. GSDS: A gene structure display server. Yi Chuan 2007, 29, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Duan, K.; Li, L.; Hu, P.; Xu, S.P.; Xu, Z.H.; Xue, H.W. A brassinolide-suppressed rice MADS-box transcription factor, OsMDP1, has a negative regulatory role in BR signaling. Plant J. 2006, 47, 519–531. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Yang, L.; Hu, W.; Zhang, J.; Xing, Y. Overexpression of an auxin receptor OsAFB6 significantly enhanced grain yield by increasing cytokinin and decreasing auxin concentrations in rice panicle. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, Q.; Wu, J.; Zhang, L.; Jiao, X.; Zhang, S.; Zhang, Z.; Sun, D.; Lu, T.; Sun, Y. Two rice authentic histidine phosphotransfer proteins, OsAHP1 and OsAHP2, mediate cytokinin signaling and stress responses in rice. Plant Physiol. 2014, 165, 335–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Cai, W.-J.; Yin, X.; Yang, D.; Dong, T.; Feng, Y.-Q.; Wu, Y. Two SLENDER AND CRINKLY LEAF dioxygenases play an essential role in rice shoot development. J. Exp. Bot. 2020, 71, 1387–1401. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Nijhawan, A.; Arora, R.; Agarwal, P.; Ray, S.; Sharma, P.; Kapoor, S.; Tyagi, A.K.; Khurana, J.P. F-box proteins in rice. Genome-wide analysis, classification, temporal and spatial gene expression during panicle and seed development, and regulation by light and abiotic stress. Plant Physiol. 2007, 143, 1467–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Niu, Q.W.; Wu, H.W.; Liu, J.; Ye, J.; Yu, N.; Chua, N.H. Analysis of non-coding transcriptome in rice and maize uncovers roles of conserved lnc RNA s associated with agriculture traits. Plant J. 2015, 84, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Xiong, L. Characterization of a Purine Permease Family Gene Os PUP 7 Involved in Growth and Development Control in Rice. J. Integr. Plant Biol. 2013, 55, 1119–1135. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Hao, Y.; Kong, D. SCARECROW has a SHORT-ROOT-independent role in modulating the sugar response1. Plant. Physiol. 2012, 158, 1769–1778. [Google Scholar] [CrossRef] [Green Version]
- Qin, H.; Wang, J.; Chen, X.; Wang, F.; Peng, P.; Zhou, Y.; Miao, Y.; Zhang, Y.; Gao, Y.; Qi, Y. Rice Os DOF 15 contributes to ethylene-inhibited primary root elongation under salt stress. New Phytol. 2019, 223, 798–813. [Google Scholar] [CrossRef]
- Yuan, J.; Wang, X.; Zhao, Y.; Khan, N.U.; Zhao, Z.; Zhang, Y.; Wen, X.; Tang, F.; Wang, F.; Li, Z. Genetic basis and identification of candidate genes for salt tolerance in rice by GWAS. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | QTLs | Lead SNPs | Chr | Position | −log10(P) | Reported Genes |
|---|---|---|---|---|---|---|
| MRL | qMRL6 | chr06_7017401 | 6 | 7017401 | 5.135 | OsPIN1a |
| qMRL8 | chr08_27868839 | 8 | 27868839 | 5.059 | OsAHP1, OsGA20ox7 | |
| TRW | qTRW3 | chr03_4592524 | 3 | 4592524 | 5.148 | OsMADS47, OsAFB6 |
| qTRW6 | chr06_6908583 | 6 | 6908583 | 5.967 | OsPIN1a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; San, M.L.; Jang, S.-G.; Lee, J.-H.; Kim, N.-E.; Lee, A.-R.; Park, S.-Y.; Cao, F.-Y.; Chin, J.-H.; Kwon, S.-W. Genome-Wide Association Study of Root System Development at Seedling Stage in Rice. Genes 2020, 11, 1395. https://doi.org/10.3390/genes11121395
Zhang H, San ML, Jang S-G, Lee J-H, Kim N-E, Lee A-R, Park S-Y, Cao F-Y, Chin J-H, Kwon S-W. Genome-Wide Association Study of Root System Development at Seedling Stage in Rice. Genes. 2020; 11(12):1395. https://doi.org/10.3390/genes11121395
Chicago/Turabian StyleZhang, Hongjia, Mar Lar San, Seong-Gyu Jang, Ja-Hong Lee, Na-Eun Kim, Ah-Rim Lee, So-Yeon Park, Fang-Yuan Cao, Joong-Hyoun Chin, and Soon-Wook Kwon. 2020. "Genome-Wide Association Study of Root System Development at Seedling Stage in Rice" Genes 11, no. 12: 1395. https://doi.org/10.3390/genes11121395
APA StyleZhang, H., San, M. L., Jang, S.-G., Lee, J.-H., Kim, N.-E., Lee, A.-R., Park, S.-Y., Cao, F.-Y., Chin, J.-H., & Kwon, S.-W. (2020). Genome-Wide Association Study of Root System Development at Seedling Stage in Rice. Genes, 11(12), 1395. https://doi.org/10.3390/genes11121395