46,XY,r(8)/45,XY,−8 Mosaicism as a Possible Mechanism of the Imprinted Birk-Barel Syndrome: A Case Study

 , , , ,
, , , ,
Abstract
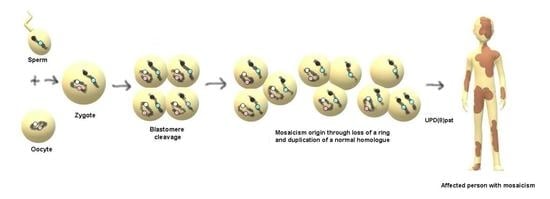
:
1. Introduction
Case Presentation
2. Materials and Methods
2.1. Materials
2.2. Cytogenetic Analyses
2.3. Array-Based Comparative Genomic Hybridization (aCGH) Analyses
2.4. Confirmation of Copy Number Variations Using Quantitative Real-Time PCR
2.5. Fluorescence In Situ Hybridization (FISH)
2.6. Targeted Massive Parallel Sequencing
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hu, Q.; Chai, H.; Shu, W.; Li, P. Human ring chromosome registry for cases in the Chinese population: Re-emphasizing cytogenomic and clinical heterogeneity and reviewing diagnostic and treatment strategies. Mol. Cytogenet. 2018, 11, 19. [Google Scholar] [CrossRef] [Green Version]
- Le Caignec, C.; Boceno, M.; Jacquemont, S.; Nguyen The Tich, S.; Rival, J.M.; David, A. Inherited ring chromosome 8 without loss of subtelomeric sequences. Ann. Genet. 2004, 47, 289–296. [Google Scholar] [CrossRef]
- Pristyazhnyuk, I.E.; Menzorov, A.G. Ring chromosomes: From formation to clinical potential. Protoplasma 2018, 255, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.; Riegel, M.; Messa, J.; Gimelli, S.; Maraschio, P.; Ciccone, R.; Stroppi, M.; Riva, P.; Perrotta, C.S.; Mattina, T.; et al. Duplications in addition to terminal deletions are present in a proportion of ring chromosomes: Clues to the mechanisms of formation. J. Med. Genet. 2008, 45, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weleber, R.G.; Verma, R.S.; Kimberling, W.J.; Fieger, H.G.; Lubs, H.A. Duplication-deficiency of the short arm of chromosome 8 following artificial insemination. Ann. Genet. 1976, 19, 241–247. [Google Scholar] [PubMed]
- Garcia-Santiago, F.A.; Martinez-Glez, V.; Santos, F.; Garcia-Minaur, S.; Mansilla, E.; Meneses, A.G.; Rosell, J.; Granero, Á.P.; Vallespín, E.; Fernández, L.; et al. Analysis of invdupdel(8p) rearrangement: Clinical, cytogenetic and molecular characterization. Am. J. Med. Genet. A 2015, 167A, 1018–1025. [Google Scholar] [CrossRef]
- Giglio, S.; Broman, K.W.; Matsumoto, N.; Calvari, V.; Gimelli, G.; Neumann, T.; Ohashi, H.; Voullaire, L.; Larizza, D.; Giorda, R.; et al. Olfactory receptor-gene clusters, genomic-inversion polymorphisms, and common chromosome rearrangements. Am. J. Hum. Genet. 2001, 68, 874–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimokawa, O.; Kurosawa, K.; Ida, T.; Harada, N.; Kondoh, T.; Miyake, N.; Yoshiura, K.; Kishino, T.; Ohta, T.; Niikawa, N.; et al. Molecular characterization of inv dup del(8p): Analysis of five cases. Am. J. Med. Genet. A 2004, 128A, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Zuffardi, O.; Bonaglia, M.; Ciccone, R.; Giorda, R. Inverted duplications deletions: Underdiagnosed rearrangements? Clin. Genet. 2009, 75, 505–513. [Google Scholar] [CrossRef]
- Sugawara, H.; Harada, N.; Ida, T.; Ishida, T.; Ledbetter, D.H.; Yoshiura, K.; Ohta, T.; Kishino, T.; Niikawa, N.; Matsumoto, N. Complex low-copy repeats associated with a common polymorphic inversion at human chromosome 8p23. Genomics 2003, 82, 238–244. [Google Scholar] [CrossRef]
- Ozgen, H.M.; van Daalen, E.; Bolton, P.F.; Maloney, V.K.; Huang, S.; Cresswell, L.; van den Boogaard, M.J.; Eleveld, M.J.; van ‘t Slot, R.; Hochstenbach, R.; et al. Copy number changes of the microcephalin 1 gene (MCPH1) in patients with autism spectrum disorders. Clin. Genet. 2009, 76, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man. Available online: http://omim.org (accessed on 11 November 2020).
- Kim, D. Physiology and pharmacology of two-pore domain potassium channels. Curr. Pharm. Des. 2005, 11, 2717–2736. [Google Scholar] [CrossRef] [PubMed]
- Chapman, C.G.; Meadows, H.J.; Godden, R.J.; Campbell, D.A.; Duckworth, M.; Kelsell, R.E.; Murdock, P.R.; Randall, A.D.; Rennie, G.I.; Gloger, I.S. Cloning, localisation and functional expression of a novel human, cerebellum specific, two pore domain potassium channel. Brain Res. Mol. Brain Res. 2000, 82, 74–83. [Google Scholar] [CrossRef]
- Zanzouri, M.; Lauritzen, I.; Duprat, F.; Mazzuca, M.; Lesage, F.; Lazdunski, M.; Patel, A. Membrane potential-regulated transcription of the resting K+ conductance TASK-3 via the calcineurin pathway. J. Biol. Chem. 2006, 281, 28910–28918. [Google Scholar] [CrossRef] [Green Version]
- Ruf, N.; Bahring, S.; Galetzka, D.; Pliushch, G.; Luft, F.C.; Nurnberg, P.; Haaf, T.; Kelsey, G.; Zechner, U. Sequence-based bioinformatic prediction and QUASEP identify genomic imprinting of the KCNK9 potassium channel gene in mouse and human. Hum. Mol. Genet. 2007, 16, 2591–2599. [Google Scholar] [CrossRef] [Green Version]
- Barel, O.; Shalev, S.A.; Ofir, R.; Cohen, A.; Zlotogora, J.; Shorer, Z.; Mazor, G.; Finer, G.; Khateeb, S.; Zilberberg, N. Maternally inherited Birk Barel mental retardation dysmorphism syndrome caused by a mutation in the genomically imprinted potassium channel KCNK9. Am. J. Hum. Genet. 2008, 83, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Graham, J.M.; Zadeh, N.; Kelley, M.; Tan, E.S.; Liew, W.; Tan, V.; Deardorff, M.A.; Wilson, G.N.; Sagi-Dain, L.; Shalev, S.A. KCNK9 imprinting syndrome-further delineation of a possible treatable disorder. Am. J. Med. Genet. A 2016, 170, 2632–2637. [Google Scholar] [CrossRef] [Green Version]
- Sediva, M.; Lassuthova, P.; Zamecnik, J.; Sedlackova, L.; Seeman, P.; Haberlova, J. Novel variant in the KCNK9 gene in a girl with Birk Barel syndrome. Eur. J. Med. Genet. 2020, 63, 103619. [Google Scholar] [CrossRef]
- Robinson, W.P.; Bottani, A.; Xie, Y.G.; Balakrishman, J.; Binkert, F.; Machler, M.; Prader, A.; Schinzel, A. Molecular, cytogenetic, and clinical investigations of Prader-Willi syndrome patients. Am. J. Hum. Genet. 1991, 49, 1219–1234. [Google Scholar]
- Pfeiffer, R.; Lenard, H. Ringchromosom 8 (46,XY,8r) bei einem debilen Jungen. Klin. Pädiatr. 1973, 185, 187–191. [Google Scholar]
- Hamers, A.J.; van Kempen, C. Ring chromosome 8 in a boy with multiple congenital abnormalities and mental retardation. J. Med. Genet. 1977, 14, 451–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mingarelli, R.; Valorani, G.; Zelante, L.; Dallapiccola, B. Ring chromosome 8 associated with microcephaly. Ann. Genet. 1991, 34, 90–92. [Google Scholar] [PubMed]
- Verma, R.S.; Conte, R.A.; Pitter, J.H.; Luke, S. Pericentric inversion of chromosome 7 (inv(7) (p22q11.2)) and ring chromosome 8 (r(8) (p23q24.3)) in a girl with minor anomalies. J. Med. Genet. 1992, 29, 66–67. [Google Scholar] [CrossRef] [PubMed]
- Bonet, H.B.; Fontenla, M.; Fauze, R.; de Pinat, G. Ring chromosome 8: Microcephaly, mental retardation and minor facial anomalies with adhesive behavioral phenotype. Rev. Neurol. 2001, 32, 746–750. [Google Scholar]
- Gradek, G.A.; Kvistad, P.H.; Houge, G. Monosomy 8 rescue gave cells with a normal karyotype in a mildly affected man with 46,XY,r(8) mosaicism. Eur. J. Med. Genet. 2006, 49, 292–297. [Google Scholar] [CrossRef]
- Database of Genomic Variants. Available online: http://projects.tcag.ca/variation (accessed on 19 November 2020).
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [Green Version]
- Zhigalina, D.I.; Skryabin, N.A.; Vasilieva, O.Y.; Lopatkina, M.E.; Vasiliev, S.A.; Sivokha, V.M.; Belyaeva, E.O.; Savchenko, R.R.; Nazarenko, L.P.; Lebedev, I.N. FISH diagnostics of chromosomal translocation with the technology of synthesis of locus-specific DNA probes based on long-range PCR. Russ. J. Genet. 2020, 56, 739–746. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data. Nucleic. Acids. Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Paez, M.T.; Yamamoto, T.; Hayashi, K.; Yasuda, T.; Harada, N.; Matsumoto, N.; Kurosawa, K.; Furutani, Y.; Asakawa, S.; Shimizu, N.; et al. Two patients with atypical interstitial deletions of 8p23.1: Mapping of phenotypical traits. Am. J. Med. Genet. A 2008, 146A, 1158–1165. [Google Scholar] [CrossRef]
- Ballarati, L.; Cereda, A.; Caselli, R.; Selicorni, A.; Recalcati, M.P.; Maitz, S.; Finelli, P.; Larizza, L.; Giardino, D. Genotype-phenotype correlations in a new case of 8p23.1 deletion and review of the literature. Eur. J. Med. Genet. 2011, 54, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Burnside, R.D.; Pappas, J.G.; Sacharow, S.; Applegate, C.; Hamosh, A.; Gadi, I.K.; Jaswaney, V.; Keitges, E.; Phillips, K.K.; Potluri, V.R.; et al. Three cases of isolated terminal deletion of chromosome 8p without heart defects presenting with a mild phenotype. Am. J. Med. Genet. A 2013, 161A, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Lin, S.; Chen, B.; Zhou, Y. Isolated chromosome 8p23.2pter deletion: Novel evidence for developmental delay, intellectual disability, microcephaly and neurobehavioral disorders. Mol. Med. Rep. 2017, 16, 6837–6845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, W.H.; Gau, S.S.; Wu, Y.Y.; Huang, Y.S.; Fang, J.S.; Chen, Y.J.; Soong, W.T.; Chiu, Y.N.; Chen, C.H. Identification and molecular characterization of two novel chromosomal deletions associated with autism. Clin. Genet. 2010, 78, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ji, T.; Wang, J.; Xiao, J.; Wang, H.; Li, J.; Gao, Z.; Yang, Y.; Cai, B.; Wang, L.; et al. Submicroscopic subtelomeric aberrations in Chinese patients with unexplained developmental delay/mental retardation. BMC Med. Genet. 2010, 11, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moog, U.; Engelen, J.J.; Albrechts, J.C.; Baars, L.G.; de Die-Smulders, C.E. Familial dup(8)(p12p21.1): Mild phenotypic effect and review of partial 8p duplications. Am. J. Med. Genet. 2000, 94, 306–310. [Google Scholar] [CrossRef]
- De Die-Smulders, C.E.; Engelen, J.J.; Schrander-Stumpel, C.T.; Govaerts, L.C.; de Vries, B.; Vles, J.S.; Wagemans, A.; Schijns-Fleuren, S.; Gillessen-Kaesbach, G.; Fryns, J.P. Inversion duplication of the short arm of chromosome 8: Clinical data on seven patients and review of the literature. Am. J. Med. Genet. 1995, 59, 369–374. [Google Scholar] [CrossRef]
- Feldman, G.L.; Weiss, L.; Phelan, M.C.; Schroer, R.J.; van Dyke, D.L. Inverted duplication of 8p: Ten new patients and review of the literature. Am. J. Med. Genet. 1993, 47, 482–486. [Google Scholar] [CrossRef]
- Minelli, A.; Floridia, G.; Rossi, E.; Clementi, M.; Tenconi, R.; Camurri, L.; Bernardi, F.; Hoeller, H.; Previde Re, C.; Maraschio, P.; et al. D8S7 is consistently deleted in inverted duplications of the short arm of chromosome 8 (inv dup 8p). Hum. Genet. 1993, 92, 391–396. [Google Scholar] [CrossRef]
- Barber, J.C.; James, R.S.; Patch, C.; Temple, I.K. Protelomeric sequences are deleted in cases of short arm inverted duplication of chromosome 8. Am. J. Med. Genet. 1994, 50, 296–299. [Google Scholar] [CrossRef]
- Guo, W.J.; Callif-Daley, F.; Zapata, M.C.; Miller, M.E. Clinical and cytogenetic findings in seven cases of inverted duplication of 8p with evidence of a telomeric deletion using fluorescence in situ hybridization. Am. J. Med. Genet. 1995, 58, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Aktas, D.; Weise, A.; Utine, E.; Alehan, D.; Mrasek, K.; von Eggeling, F.; Thieme, H.; Tuncbilek, E.; Liehr, T. Clinically abnormal case with paternally derived partial trisomy 8p23.3 to 8p12 including maternal isodisomy of 8p23.3: A case report. Mol. Cytogenet. 2009, 2, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ChromosOmics Database. Available online: http://cs-tl.de/DB.html (accessed on 19 November 2020).
- Bershteyn, M.; Hayashi, Y.; Desachy, G.; Hsiao, E.C.; Sami, S.; Tsang, K.M.; Weiss, L.A.; Kriegstein, A.R.; Yamanaka, S.; Wynshaw-Boris, A. Cell-autonomous correction of ring chromosomes in human induced pluripotent stem cells. Nature 2014, 507, 99–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gridina, M.M.; Nikitina, T.V.; Orlova, P.A.; Minina, J.M.; Kashevarova, A.A.; Yakovleva, Y.S.; Lopatkina, M.E.; Vasilyev, S.A.; Fedotov, D.A.; Mikhailik, L.I.; et al. Establishment of an induced pluripotent stem cell line (ICGi025-A) from fibroblasts of a patient with 46,XY,r(8)/45,XY,−8 mosaicism. Stem Cell Res. 2020, 49, 1020204. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pfeiffer and Lenard [21] | Hamers and van Kempen [22] | Mingarelli et al. [23] | Verma et al. [24] | Bonet et al. [25] | Le Caignec et al. [2] Patient III-1, 6.5-Year-Old Boy | Le Caignec et al. [2] Patient II-1, Mother of Patient III-1, 30 Years Old | Gradek et al. [26] | Index Patient, This Study | |
|---|---|---|---|---|---|---|---|---|---|
| Karyotype | 46,XY,r(8)/ 46,XY? | 46,XY,r(8)[117]/ 46,XY[1]/ 45,XY,−8[1]/ 47,XY,r(8),+r(8) [1]—lymphocytes 46,XY[64]/47,XY,r(8),+r(8) [5]/?[3]—fibroblasts | 46,XX,r(8) | 46,XX, r(8), inv(7) | 46,XY,r(8)/ 45,XY,−8/ 47,XY,r(8), +r(8) | 46,XY,r(8)(p23q24.3) [24]/45,XY,−8[2].ish r(8)(8ptel+,8qtel+) | 46,XX,r(8)(p23q24.3) [22]/45,XX,−8[2]/ 47,XX,r(8)(p23q24.3), +r(8)(p23q24.3)[1].ish r(8)(8ptel+, 8qtel+) | 9 years, lymphocytes: 46,XY,r(8)[27]/ 46,XY, tan r(8)[1]/ 45,XY,−8[2]; 12 years, lymphocytes: 46,XY,r(8)[27]/ 46,XY, tan r(8)[2]/46,XY[1]; 19 years, lymphocytes: 46,XY,r(8)[23]/ 46,XY[7]; 12 years, skin fibroblasts: 46,XY,r(8)[29]/45,XY,−8[3]/ 46,XY[18] | 1 year 4 months, lymphocytes: 46,XY,r(8)(p23q24.3)[27]/ 45,XY,−8[3] |
| Ring origin | − | − | − | − | − | Maternal | Maternal | Maternal | Maternal |
| Family history | − | No abortions or stillbirths | − | − | − | − | No previous history of miscarriage | Two normal siblings | First pregnancy ended with miscarriage |
| Intrauterine growth retardation | − | − | − | − | − | + | − | − | − |
| Birth weight | − | 2770 g (10th centile) | − | 2000 g (<3rd centile) | − | 1350 g (5th centile) | − | 3170 g (25th centile) | 2720 g (3–10th centiles) |
| Body length | − | 47 cm (10th centile) | − | − | − | 38.5 cm (5th centile) | − | 50 cm (50th centile) | 48 cm (10th centile) |
| Head circumference | − | − | − | 29 cm (<3rd centile) | − | 27.5 cm (<3rd centile) | − | 33 cm (5–10th centiles) | 31 cm (3rd centile) |
| Gestational age at birth | − | (13 days after term) | − | 40 weeks | − | Premature | − | At term | 38 weeks |
| Developmental delay | − | + | − | + | − | + | − | + | + |
| Short stature | + | + | + | − | + | + | + | + | − |
| Microcephaly | + | + | + | + | + | + | + | + | + |
| Facial dysmorphia | Turricephaly, flat occiput, hypotelorism, dental anomalies, micrognathia | Dolichocephaly, prominent occiput, bilateral strabismus, epicanthic folds, asymmetric ears, thin upper lip, gothic palate, asymmetry of the upper dental arch, micrognathia | Slightly sloping forehead, flat face with prominent glabella, upward slanted palpebral fissures, hypertelorism, bilateral epicanthic folds, flat nasal bridge | Prominent nose, high arched palate, low-set left ear | Hypotelorism, bilateral epicanthic folds, long philtrum, narrow palate, low-set ears, thin lips, micrognathia | Small nose, anteverted nostrils, long philtrum, thin upper lip | − | Brachycephaly, antimongoloid slant, bilateral epicanthus, prominent ears | High forehead, flat sloping occiput, short neck, abnormal hairline, preauricular tag of the left ear, bulbous nose, smooth philtrum, macrostomy, thick lips, high palate, irregular teeth growth, micrognathia |
| Other anomalies | Unilateral cryptorchidism, coxa valga | Clinobrachydactyly of fifth fingers, pectus excavatum, scapulae alatae, long thorax, amblyopy, camptodactyly of both fifth fingers, hernia inguinalis (bilateral and operated), dislocation of the hip, hypertelorism of the nipples, sacral dimples, dimples dorsal of the elbows, cutis marmorata | Brachydactyly of the fifth fingers | Bilateral equinovarus feet, limitation of dorsiflexion of the feet and joints, hyperreflexia | Clinobrachydactyly of the fifth fingers | Clinobrachydactyly of the fifth fingers, amblyopy | − | Broad neck, hypertelorism of the nipples, mild brachydactyly of the fifth fingers | Broad fingers, proximally placed thumb (bilateral), thickening of the distal phalanx of the hallux (bilateral), dysplastic nails, pectus excavatum, pes planovalgus |
| Speech and language delay | − | − | − | No speech at 13 years | − | + | − | + | + |
| Learning difficulties | − | − | − | − | − | + | Mild | + | n/a |
| Intellectual disability | Mild | Severe | Mild | Severe | Moderate | Mild | Normal intelligence | Borderline to mild (IQ 70) | n/a |
| Behavior | − | − | Hyperactivity, pleasant personality | Hyperactivity, self-stimulatory behavior, no social contact, unable to feed herself | Pleasant personality, attachment for people and things with unrestricted affect | ADHD | − | ADHD, kind, confident, empathic, socially well adjusted | − |
| MRI, CT, echoencephalography | − | − | − | − | − | Focal pachygyria with decreased sulcation and a thickened cortex in the frontal and occipital areas bilaterally, mild-amplitude rhythmic activity | − | Normal | Ventriculomegaly |
| Feeding | − | Difficulty in the neonatal period | − | − | − | − | − | Regurgitation, vomiting, poor appetite | Mild dysphagia |
| Seizures | − | + | − | − | − | − | − | − | − |
| Recurrent infections | − | + | − | − | − | − | − | − | − |
| Hypotonia | − | + | − | − | − | − | − | + | + |
| Decreased fetal movement during the pregnancy | − | − | − | + | − | − | − | − | − |
| Constipation | − | − | − | − | − | − | − | + | − |
| Barel et al. [17] 15 Members of the Family | Graham et al. [18] Patient 1 | Graham et al. [18] Patient 2 | Graham et al. [18] Patient 3 | Graham et al. [18] Patient 4 | Sevida et al. [19] | |
|---|---|---|---|---|---|---|
| Mutation | Missense mutation 770G>A in exon 2, replacing glycine at position 236 by arginine (G236R), in KCNK9 | De novo c.706G>C mutation (pGly236Arg) in KCNK9 | De novo c.706G >C mutation (pGly236Arg) in KCNK9 | De novo c.706G>C mutation (pGly236Arg) in KCNK9 | De novo heterozygous c.706G>A; p.G236R mutation in the KCNK9 gene and m.8902G>A; pA126T variant of uncertain significance in the mitochondrial ATP6 gene | Heterozygous c.710C>A (p.A237D) in KCNK9 |
| Gestational age at birth | − | At term | At term | At term | 38.5 weeks | At term |
| Birth weight | − | 2558 g (3–5th centile) | 2655 g (10–25th centiles) | 2954 g (10–25th centiles) | 3302 g (50th centile) | 3210 g (25th centile) |
| Birth length | − | 43.2 cm (<3rd centile) | 48 cm (25th centile) | 53 cm (>90th centile) | 52 cm (80th centile) | 50 cm (50th centile) |
| Head circumference | − | 33.2 cm (10–25th centiles) | 35 cm (50th centile) | 34.5 cm (75th centile) | 34.5 cm (25th centile) | 53.5 cm at 13 years * (50th centile) |
| Oligohydramnios | − | + | − | − | − | − |
| Intrauterine growth restriction | − | + | − | − | − | − |
| Hypoglycemia | − | + | − | − | − | − |
| Developmental delay | − | + | + | + | + | + |
| Intelligence | Moderate to severe intellectual disability | − | − | − | − | Border-line intellectual deficit |
| Behavior and sleep | Hyperactive | Lethargic, fussy, and uncommunicative, sleeping 13–14 h a day | Severe obstructive sleep apnea with both central and obstructive patterns | Fatigability | Interacting well with other people | |
| Feeding | Severe difficulties in infancy (tube feeding), dysphagia of solid foods until near puberty | Severe feeding problems requiring a gastrostomy tube due to poor sucking and gastroesophageal reflux | Moderate oropharyngeal dysphagia with silent aspiration of thin fluids that required nasogastric tube feedings | Poor feeding | Poor feeding | Weak sucking and episodes of hypoxia during feeding in infancy, dysphagia of liquids with a well-evocable pharyngeal reflex at 17 years |
| Muscle tone | Generalized hypotonia at an early age followed by weakness of proximal muscles and of the supra- and infrascapular and trapezius muscles later on | Central hypotonia with episodes of spontaneous clonus upon awakening | Congenital hypotonia, diminished facial movements, myoclonic jerks, proximal muscle weakness | Generalized hypotonia, weakness | Mild generalized hypotonia | Generalized muscle weakness, mild cerebellar syndrome, peripheral motoneuron syndrome, areflexia, problems with fine and gross motor skills, mild contractures of triceps surae muscles and decreased physical endurance, hypomimia, bilateral lagophthalmos, tongue fasciculations |
| Facial dysmorphia | Elongated face with a narrow bitemporal diameter, mild atrophy of the temporalis and masseter muscles, reduced facial movements; flared, bushy, and arched upward eyebrows, downturned eyelids, sparse eyelashes in the inner third of the lower eyelids, congested conjunctivae; protruding ears with a very prominent fold of the crux of the helix and a prominent antihelical fold; high and narrow nasal bridge with a broad nasal tip; extremely short, broad, and thick philtrum; prominent maxillary and premaxillary regions, hypotonia of the mandible, micrognathia, open mouth; thick lips, downturned upper lip (“fish mouth”), a lower lip shorter than the upper lip; narrow, high-arched palate with a full or submucous cleft; large and protruding incisors | Dolichocephaly with bitemporal narrowing, upswept anterior and posterior hair pattern, short philtrum, tented upper lip, V-shaped cleft palate, prominent maxilloalveolar frenulum, small mandible, medially flared eyebrows | Bitemporal narrowing, tented upper lip, high arched palate, and retrognathia | Thin upper lip, downturned open mouth, broad alveolar ridges, cleft soft palate, micrognathia/retrognathia | A high and broad nasal bridge, a generous mouth with downturned corners | Slightly elongated face, cleft palate, micrognathia, tented upper lip, short philtrum, low-set ears |
| Other abnormalities | Narrow, elongated neck, trunk, and feet; mild joint contractures of the hips, elbows, phalanx, and feet; pilonidal dimple or sinus | Tapered fingers with prominent fetal fingertip pads, bridged transverse palmar flexion creases, joint laxity | Sacral dimple, extradural lipoma | Patent foramen ovale, sacral dimple, phimosis, small thumbs, soft doughy hands, small feet with high arches, dorsiflexed toes, tremor-like movements of the arms with intention and mild head bobbing, especially when tired | Dextroscoliosis in the thoracic region of 10–15° | − |
| Speech | Dysphonic | A word, “mama”, at age 19 months under treatment with mefenamic acid | Mainly vowels | − | Several words | Dysarthria, dysphonia |
| Electromyography and muscle biopsy | Muscle biopsy: compatible with spinal muscular atrophy | − | Electromyography of the right biceps and quadriceps suggested a generalized myopathy, and muscle biopsy showed mild variation in fiber size with a few perivascular mononuclear inflammatory infiltrates. Immunostaining with monoclonal antibodies to sarcoglycans revealed diffuse, slightly reduced sarcolemmal staining of delta-sarcoglycan, patchy reduction in β-sarcoglycan | Normal muscle ultrasound | − | EMG revealed signs of primarily axonal peripheral pure motor neuropathy. The pattern on EMG was similar to the pattern observed in spinal muscular atrophy. A muscle biopsy performed at the age of 18 months showed signs of neurogenic transformation with mosaic selective atrophy of type 2 fibbers and hypertrophy of type 1 fibers |
| Hyperinsulinism | − | + | − | − | − | − |
| Markedly diminished tearing upon crying | − | + | + | + | − | − |
| Clinical Features of Birk-Barel Syndrome | Patients with Maternal r(8), [2,26] | Index Patient |
|---|---|---|
| Low weight at birth | + | + |
| Short length at birth | + | + |
| Microcephaly at birth | + | + |
| Developmental delay/intellectual disability | + | + |
| Elongated face | − | + |
| Bushy eyebrows | − | + |
| Long eyelashes | − | + |
| Conjunctivitis | − | + |
| Abnormal ears | + | + |
| Short philtrum | − | + |
| Micrognathia | − | + |
| Feeding problems | + | + |
| Reduced muscle tone | + | + |
| Restricted facial movements | − | + |
| Abnormal speech | + | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kashevarova, A.A.; Nikitina, T.V.; Mikhailik, L.I.; Belyaeva, E.O.; Vasilyev, S.A.; Lopatkina, M.E.; Fedotov, D.A.; Fonova, E.A.; Zarubin, A.A.; Sivtsev, A.A.; et al. 46,XY,r(8)/45,XY,−8 Mosaicism as a Possible Mechanism of the Imprinted Birk-Barel Syndrome: A Case Study. Genes 2020, 11, 1473. https://doi.org/10.3390/genes11121473
Kashevarova AA, Nikitina TV, Mikhailik LI, Belyaeva EO, Vasilyev SA, Lopatkina ME, Fedotov DA, Fonova EA, Zarubin AA, Sivtsev AA, et al. 46,XY,r(8)/45,XY,−8 Mosaicism as a Possible Mechanism of the Imprinted Birk-Barel Syndrome: A Case Study. Genes. 2020; 11(12):1473. https://doi.org/10.3390/genes11121473
Chicago/Turabian StyleKashevarova, Anna A., Tatyana V. Nikitina, Larisa I. Mikhailik, Elena O. Belyaeva, Stanislav A. Vasilyev, Mariya E. Lopatkina, Dmitry A. Fedotov, Elizaveta A. Fonova, Aleksei A. Zarubin, Aleksei A. Sivtsev, and et al. 2020. "46,XY,r(8)/45,XY,−8 Mosaicism as a Possible Mechanism of the Imprinted Birk-Barel Syndrome: A Case Study" Genes 11, no. 12: 1473. https://doi.org/10.3390/genes11121473
APA StyleKashevarova, A. A., Nikitina, T. V., Mikhailik, L. I., Belyaeva, E. O., Vasilyev, S. A., Lopatkina, M. E., Fedotov, D. A., Fonova, E. A., Zarubin, A. A., Sivtsev, A. A., Skryabin, N. A., Nazarenko, L. P., & Lebedev, I. N. (2020). 46,XY,r(8)/45,XY,−8 Mosaicism as a Possible Mechanism of the Imprinted Birk-Barel Syndrome: A Case Study. Genes, 11(12), 1473. https://doi.org/10.3390/genes11121473