Distinct Effects of Inflammation on Cytochrome P450 Regulation and Drug Metabolism: Lessons from Experimental Models and a Potential Role for Pharmacogenetics



Abstract
:1. Introduction
2. Effects of Inflammatory Stimuli on CYP Expression Levels and Activity in Human In-Vitro Liver Models
2.1. Interleukin-6 (IL-6)
2.1.1. Maximal Effect (Emax) (mRNA)
2.1.2. Sensitivity between CYPs (mRNA)
2.1.3. Sensitivity between PHH Donors
2.1.4. Drug-Metabolizing Activity
2.1.5. Pathways
2.1.6. Long-Term Studies
2.1.7. Clinic
2.2. Interleukin 1 (IL-1)-Family: Interleukin-1β and Interleukin-18
2.2.1. Maximal Effect (Emax) (mRNA)
2.2.2. Sensitivity between Models
2.2.3. Sensitivity between PHH Donors
2.2.4. Pathways
2.3. Tumor Necrosis Factor α (TNF-α)
2.4. Pathogen Associated Molecular Patterns (PAMPs)
2.5. Other Cytokines: Transforming Growth Factor β (TGF-β), Interferon γ (IFN-γ), Interleukin-22 (IL-22), Interleukin-23 (IL-23), and Interleukin-2 (IL-2)
2.6. Summary
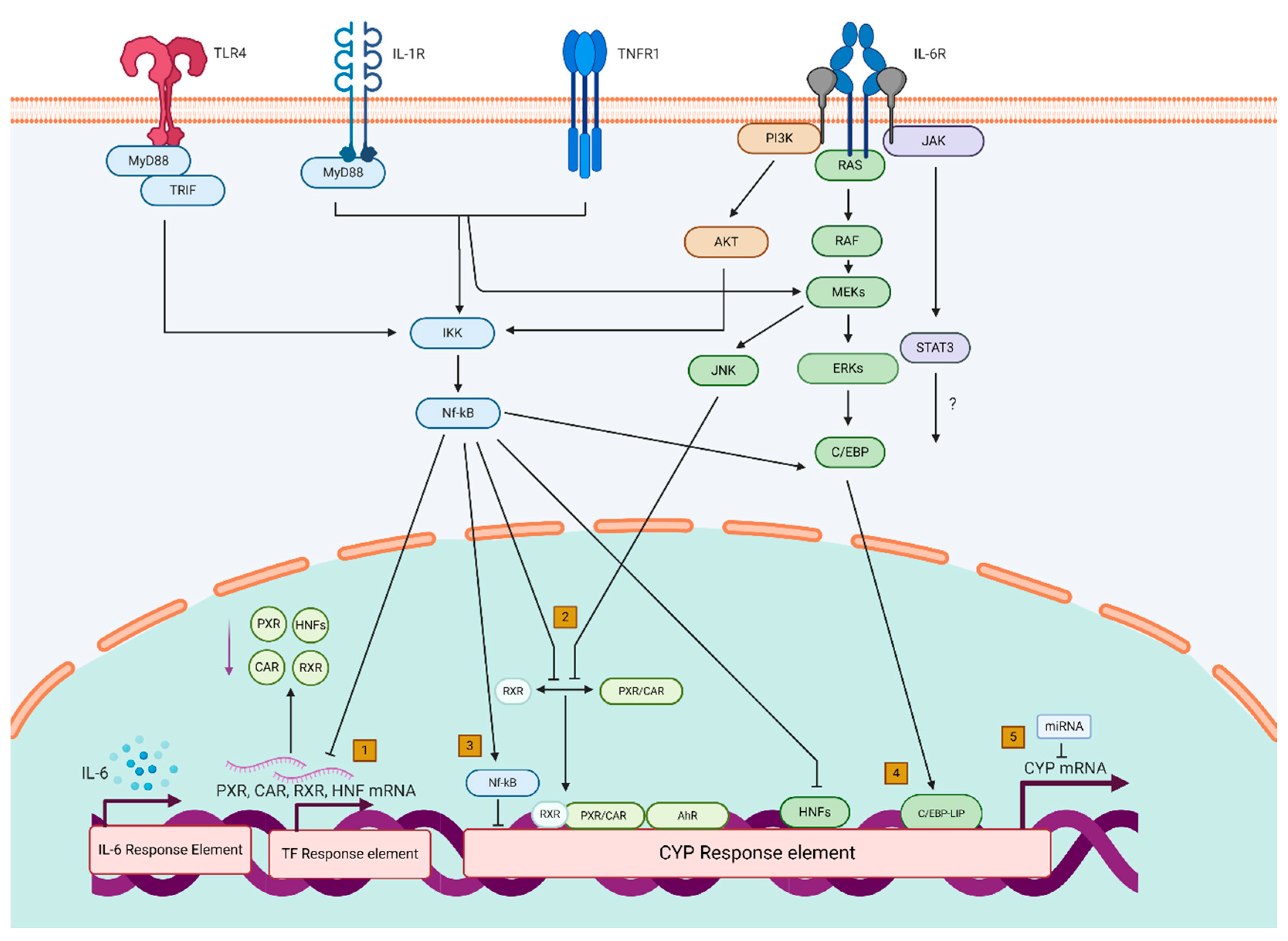
3. Mechanistic Pathways via Which Inflammation Modulates Hepatic Functions That Are Critical for Drug Metabolism
3.1. Transcriptional Downregulation of Transcription Factors
3.1.1. Downregulation of Nuclear Receptors
3.1.2. Downregulation of Hepatocyte Nuclear Factors
3.2. Interference with Dimerization/Nuclear Translocation of (Nuclear) Transcription Factors
3.3. Direct Regulation by NF-κB
3.4. Altered Liver-Enriched C/EBP Signaling
3.5. Posttranscriptional Mechanisms (miRNA)
3.6. Concluding Remark
4. Pharmacogenetic Variation in Inflammatory Pathways and the Effect on Drug Pharmacokinetics
4.1. Genetic Variation: Inflammatory Mediators
4.2. Genetic Variation: Inflammatory Receptors
4.3. Genetic Variation: Inflammatory Transcription Factors (NF-κB)
4.4. Genetic Variation: Nuclear Receptors (PXR, CAR)
4.5. Genetic Variation: Cytochrome P450 Enzymes
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wilkinson, G.R. Drug therapy: Drug metabolism and variability among patients in drug response. N. Engl. J. Med. 2005, 352, 2211–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.R.; Smith, R.L. Inflammation-induced phenoconversion of polymorphic drug metabolizing enzymes: Hypothesis with implications for personalized medicine. Drug Metab. Dispos. 2015, 43, 400–410. [Google Scholar] [CrossRef] [Green Version]
- Stanke-Labesque, F.; Gautier-Veyret, E.; Chhun, S.; Guilhaumou, R. Inflammation is a major regulator of drug metabolizing enzymes and transporters: Consequences for the personalization of drug treatment. Pharmacol. Ther. 2020. [Google Scholar] [CrossRef]
- Vet, N.J.; Brussee, J.M.; De Hoog, M.; Mooij, M.G.; Verlaat, C.W.M.; Jerchel, I.S.; Schaik, R.H.N.V.; Koch, B.C.P.; Tibboel, D.; Knibbe, C.A.J.; et al. Inflammation and organ failure severely affect midazolam clearance in critically ill children. Am. J. Respir. Crit. Care Med. 2016, 194, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Van Wanrooy, M.J.P.; Span, L.F.R.; Rodgers, M.G.G.; Van Heuvel, E.R.D.; Uges, D.R.A.; Van Werf, T.S.D.; Kosterink, J.G.W.; Alffenaara, J.W.C. Inflammation is associated with voriconazole trough concentrations. Antimicrob. Agents Chemother. 2014, 58, 7098–7101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, Y.; de Vries, D.E.; Xu, Z.; Marciniak, S.J.; Chen, D.; Leon, F.; Davis, H.M.; Zhou, H. Evaluation of disease-mediated therapeutic protein-drug interactions between an anti-interleukin-6 monoclonal antibody (sirukumab) and cytochrome P450 activities in a phase 1 study in patients with rheumatoid arthritis using a cocktail approach. J. Clin. Pharmacol. 2015, 55, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.D.; Bansal, A.; Hassman, D.; Akinlade, B.; Li, M.; Li, Z.; Swanson, B.; Hamilton, J.D.; DiCioccio, A.T. Evaluation of Potential Disease-Mediated Drug–Drug Interaction in Patients with Moderate-to-Severe Atopic Dermatitis Receiving Dupilumab. Clin. Pharmacol. Ther. 2018, 104, 1146–1154. [Google Scholar] [CrossRef]
- Gravel, S.; Chiasson, J.L.; Turgeon, J.; Grangeon, A.; Michaud, V. Modulation of CYP450 Activities in Patients With Type 2 Diabetes. Clin. Pharmacol. Ther. 2019, 106, 1280–1289. [Google Scholar] [CrossRef]
- Sim, S.C.; Kacevska, M.; Ingelman-Sundberg, M. Pharmacogenomics of drug-metabolizing enzymes: A recent update on clinical implications and endogenous effects. Pharm. J. 2013, 13, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Aitken, A.E.; Richardson, T.A.; Morgan, E.T. Regulation of drug-metabolizing enzymes and transporters in inflammation. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 123–149. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, F. Potential pharmacokinetic interactions of therapeutic cytokines or cytokine modulators on small-molecule drugs: Mechanistic understanding via studies using in vitro systems. Drug Metabol. Drug Interact. 2014, 29, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Martignoni, M.; Groothuis, G.M.M.; de Kanter, R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2006, 2, 875–894. [Google Scholar] [CrossRef] [PubMed]
- Aitken, A.E.; Morgan, E.T. Gene-specific effects of inflammatory cytokines on cytochrome P450 2C, 2B6 and 3A4 mRNA levels in human hepatocytes. Drug Metab. Dispos. 2007, 35, 1687–1693. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.; Thomas, M.; Hofmann, U.; Seehofer, D.; Damm, G.; Zanger, U.M. A systematic comparison of the impact of inflammatory signaling on absorption, distribution, metabolism, and excretion gene expression and activity in primary human hepatocytes and HepaRG Cells. Drug Metab. Dispos. 2015, 43, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Febvre-James, M.; Bruyère, A.; Le Vée, M.; Fardel, O. The jak1/2 inhibitor ruxolitinib reverses interleukin-6-mediated suppression of drug-detoxifying proteins in cultured human hepatocytes. Drug Metab. Dispos. 2018, 46, 131–140. [Google Scholar] [CrossRef]
- Rubin, K.; Janefeldt, A.; Andersson, L.; Berke, Z.; Grime, K.; Andersson, T.B. HepaRG cells as human-relevant in vitro model to study the effects of inflammatory stimuli on cytochrome P450 isoenzymes. Drug Metab. Dispos. 2015, 43, 119–125. [Google Scholar] [CrossRef]
- Dallas, S.; Sensenhauser, C.; Batheja, A.; Singer, M.; Markowska, M.; Zakszewski, C.; Mamidi, R.N.V.S.; McMillia, M.; Han, C.; Zhou, H.; et al. De-Risking Bio-therapeutics for Possible Drug Interactions Using Cryopreserved Human Hepatocytes. Curr. Drug Metab. 2012, 13, 923–929. [Google Scholar] [CrossRef]
- Dickmann, L.J.; Patel, S.K.; Rock, D.A.; Wienkers, L.C.; Slatter, J.G. Effects of interleukin-6 (IL-6) and an anti-IL-6 monoclonal antibody on drug-metabolizing enzymes in human hepatocyte culture. Drug Metab. Dispos. 2011, 39, 1415–1422. [Google Scholar] [CrossRef] [Green Version]
- Sunman, J.A.; Hawke, R.L.; LeCluyse, E.L.; Kashuba, A.D.M. Kupffer cell-mediated IL-2 suppression of CYP3A activity in human hepatocytes. Drug Metab. Dispos. 2004, 32, 359–363. [Google Scholar] [CrossRef] [Green Version]
- Long, T.J.; Cosgrove, P.A.; Dunn, R.T.; Stolz, D.B.; Hamadeh, H.; Afshari, C.; McBride, H.; Griffith, L.G. Modeling therapeutic antibody-small molecule drug-drug interactions using a three-dimensional perfusable human liver coculture platform. Drug Metab. Dispos. 2016, 44, 1940–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.V.; Ukairo, O.; Khetani, S.R.; McVay, M.; Kanchagar, C.; Seghezzi, W.; Ayanoglu, G.; Irrechukwu, O.; Evers, R. Establishment of a hepatocyte-kupffer cell coculture model for assessment of proinflammatory cytokine effects on metabolizing enzymes and drug transporters. Drug Metab. Dispos. 2015, 43, 774–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, N.; Kubik, L.; Luckert, C.; Thomas, M.; Hofmann, U.; Zanger, U.M.; Böhmert, L.; Lampen, A.; Braeuning, A. Regulation of drug metabolism by the interplay of inflammatory signaling, steatosis, and xeno-sensing receptors in HepaRG cells. Drug Metab. Dispos. 2018, 46, 326–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickmann, L.J.; Patel, S.K.; Wienkers, L.C.; Greg Slatter, J. Effects of Interleukin 1β (IL-1β) and IL-1β/Interleukin 6 (IL-6) Combinations on Drug Metabolizing Enzymes in Human Hepatocyte Culture. Curr. Drug Metab. 2012, 13, 930–937. [Google Scholar] [CrossRef]
- Le Vée, M.; Bruyère, A.; Jouan, E.; Fardel, O. Janus kinase-dependent regulation of drug detoxifying protein expression by interleukin-22 in human hepatic cells. Int. Immunopharmacol. 2020, 83, 106439. [Google Scholar] [CrossRef]
- Dallas, S.; Chattopadhyay, S.; Sensenhauser, C.; Batheja, A.; Singer, M.; Silva, J. Interleukins-12 and -23 do not alter expression or activity of multiple cytochrome p450 enzymes in cryopreserved human hepatocytes. Drug Metab. Dispos. 2013, 41, 689–693. [Google Scholar] [CrossRef] [Green Version]
- Taub, R. Liver regeneration: From myth to mechanism. Nat. Rev. Mol. Cell Biol. 2004, 5, 836–847. [Google Scholar] [CrossRef]
- Han, H.; Ma, Q.; Li, C.; Liu, R.; Zhao, L.; Wang, W.; Zhang, P.; Liu, X.; Gao, G.; Liu, F.; et al. Profiling serum cytokines in COVID-19 patients reveals IL-6 and IL-10 are disease severity predictors. Emerg. Microbes Infect. 2020, 9, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Robak, T.; Gladalska, A.; Stepień, H.; Robak, E. Serum levels of interleukin-6 type cytokines and soluble interleukin-6 receptor in patients with rheumatoid arthritis. Mediat. Inflamm. 1998, 7, 347–353. [Google Scholar] [CrossRef]
- Evers, R.; Dallas, S.; Dickmann, L.J.; Fahmi, O.A.; Kenny, J.R.; Kraynov, E.; Nguyen, T.; Patel, A.H.; Slatter, J.G.; Zhang, L. Critical review of preclinical approaches to investigate cytochrome P450-mediated therapeutic protein drug-drug interactions and recommendations for best practices: A white paper. Drug Metab. Dispos. 2013, 41, 1598–1609. [Google Scholar] [CrossRef]
- Dluzen, D.F.; Lazarus, P. MicroRNA regulation of the major drug-metabolizing enzymes and related transcription factors. Drug Metab. Rev. 2015, 47, 320–334. [Google Scholar] [PubMed]
- Reeh, H.; Rudolph, N.; Billing, U.; Christen, H.; Streif, S.; Bullinger, E.; Schliemann-Bullinger, M.; Findeisen, R.; Schaper, F.; Huber, H.J.; et al. Response to IL-6 trans- A nd IL-6 classic signalling is determined by the ratio of the IL-6 receptor α to gp130 expression: Fusing experimental insights and dynamic modelling. Cell Commun. Signal. 2019, 17, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, R.; Klein, M.; Thomas, M.; Dräger, A.; Metzger, U.; Templin, M.F.; Joos, T.O.; Thasler, W.E.; Zell, A.; Zanger, U.M. Coordinating Role of RXRα in Downregulating Hepatic Detoxification during Inflammation Revealed by Fuzzy-Logic Modeling. PLoS Comput. Biol. 2016, 12, e1004431. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Antona, C.; Donato, M.T.; Boobis, A.; Edwards, R.J.; Watts, P.S.; Castell, J.V.; Gómez-Lechón, M.J. Cytochrome P450 expression in human hepatocytes and hepatoma cell lines: Molecular mechanisms that determine lower expression in cultured cells. Xenobiotica 2002, 32, 505–520. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef]
- Parent, R.; Marion, M.J.; Furio, L.; Trépo, C.; Petit, M.A. Origin and Characterization of a Human Bipotent Liver Progenitor Cell Line. Gastroenterology 2004, 126, 1147–1156. [Google Scholar] [CrossRef]
- Szabo, G.; Petrasek, J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387–400. [Google Scholar] [CrossRef]
- Bachmann, M.; Pfeilschifter, J.; Mühl, H. A prominent role of interleukin-18 in acetaminophen-induced liver injury advocates its blockage for therapy of hepatic necroinflammation. Front. Immunol. 2018, 9, 161. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Xu, M.-J.; Gao, B. Hepatocytes: A key cell type for innate immunity. Cell. Mol. Immunol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Cahill, C.M.; Rogers, J.T. Interleukin (IL) 1β induction of IL-6 is mediated by a novel phosphatidylinositol 3-kinase-dependent AKT/IκB kinase α pathway targeting activator protein-1. J. Biol. Chem. 2008, 283, 25900–25912. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.H.; Sun, S.C. Tumor necrosis factor receptor-associated factor regulation of nuclear factor κB and mitogen-activated protein kinase pathways. Front. Immunol. 2018, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Uppal, H.; Saini, S.P.S.; Mu, Y.; Little, J.M.; Radominska-Pandya, A.; Zemaitis, M.A. Orphan nuclear receptor-mediated xenobiotic regulation in drug metabolism. Drug Discov. Today 2004, 9, 442–449. [Google Scholar] [CrossRef]
- Kojima, K.; Nagata, K.; Matsubara, T.; Yamazoe, Y. Broad but distinct role of pregnane x receptor on the expression of individual cytochrome p450s in human hepatocytes. Drug Metab. Pharmacokinet. 2007, 22, 276–286. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Ferguson, S.S.; Negishi, M.; Goldstein, J.A. Identification of constitutive androstane receptor and glucocorticoid receptor binding sites in the CYP2C19 promoter. Mol. Pharmacol. 2003, 64, 316–324. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, S.S.; Lecluyse, E.L.; Negishi, M.; Goldstein, J.A. Regulation of human CYP2C9 by the constitutive androstane receptor: Discovery of a new distal binding site. Mol. Pharmacol. 2002, 62, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Barwick, J.L.; Simon, C.M.; Pierce, A.M.; Safe, S.; Blumberg, B.; Guzelian, P.S.; Evans, R.M. Reciprocal activation of xenobiotic response genes by nuclear receptors SXR/PXR and CAR. Genes Dev. 2000, 14, 3014–3023. [Google Scholar] [CrossRef] [Green Version]
- Pascussi, J.M.; Gerbal-Chaloin, S.; Pichard-Garcia, L.; Daujat, M.; Fabre, J.M.; Maurel, P.; Vilarem, M.J. Interleukin-6 negatively regulates the expression of pregnane X receptor and constitutively activated receptor in primary human hepatocytes. Biochem. Biophys. Res. Commun. 2000, 274, 707–713. [Google Scholar] [CrossRef]
- Sun, H.Y.; Yan, Y.J.; Li, Y.H.; Lv, L. Reversing effects of ginsenosides on LPS-induced hepatic CYP3A11/3A4 dysfunction through the pregnane X receptor. J. Ethnopharmacol. 2019, 229, 246–255. [Google Scholar] [CrossRef]
- Beigneux, A.P.; Moser, A.H.; Shigenaga, J.K.; Grunfeld, C.; Feingold, K.R. Reduction in cytochrome P-450 enzyme expression is associated with repression of CAR (constitutive androstane receptor) and PXR (pregnane X receptor) in mouse liver during the acute phase response. Biochem. Biophys. Res. Commun. 2002, 293, 145–149. [Google Scholar] [CrossRef]
- Yang, J.; Hao, C.; Yang, D.; Shi, D.; Song, X.; Luan, X.; Hu, G.; Yan, B. Pregnane X receptor is required for interleukin-6-mediated down-regulation of cytochrome P450 3A4 in human hepatocytes. Toxicol. Lett. 2010, 197, 219–226. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Li, X.; Wang, X.; Jin, X.; Shi, D.; Wang, J.; Bi, D. Cecropin B represses CYP3A29 expression through activation of the TLR2/4-NF-κ B/PXR signaling pathway. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Assenat, E.; Gerbal-Chaloin, S.; Larrey, D.; Saric, J.; Fabre, J.M.; Maurel, P.; Vilarem, M.J.; Pascussi, J.M. Interleukin 1β inhibits CAR-induced expression of hepatic genes involved in drug and bilirubin clearance. Hepatology 2004, 40, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Richardson, T.A.; Morgan, E.T. Hepatic cytochrome P450 gene regulation during endotoxin-induced inflammation in nuclear receptor knockout mice. J. Pharmacol. Exp. Ther. 2005, 314, 703–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, I.; Bresnick, E. Regulation of the constitutive expression of the human CYP1A2 gene: Cis elements and their interactions with proteins. Mol. Pharmacol. 1995, 47, 677–685. [Google Scholar]
- Kamiyama, Y.; Matsubara, T.; Yoshinari, K.; Nagata, K.; Kamimura, H.; Yamazoe, Y. Role of Human Hepatocyte Nuclear Factor 4α in the Expression of Drug-Metabolizing Enzymes and Transporters in Human Hepatocytes Assessed by Use of Small Interfering RNA. Drug Metab. Pharmacokinet. 2007, 22, 287–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirona, R.G.; Lee, W.; Leake, B.F.; Lan, L.B.; Cline, C.B.; Lamba, V.; Parviz, F.; Duncan, S.A.; Inoue, Y.; Gonzalez, F.J.; et al. The orphan nuclear receptor HNF4α determines PXR- and CAR-mediated xenobiotic induction of CYP3A4. Nat. Med. 2003, 9, 220–224. [Google Scholar] [CrossRef]
- Martínez-Jiménez, C.P.; Castell, J.V.; Gómez-Lechón, M.J.; Jover Atienza, R. Transcriptional activation of CYP2C9, CYP1A1, and CYP1A2 by hepatocyte nuclear factor 4α requires coactivators peroxisomal proliferator activated receptor-γ coactivator 1α and steroid receptor coactivator 1. Mol. Pharmacol. 2006, 70, 1681–1692. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, S.S.; Chen, Y.; LeCluyse, E.L.; Negishi, M.; Goldstein, J.A. Human CYP2C8 is transcriptionally regulated by the nuclear receptors constitutive androstane receptor, pregnane X receptor, glucocorticoid receptor, and hepatic nuclear factor 4α. Mol. Pharmacol. 2005, 68, 747–757. [Google Scholar] [CrossRef]
- Cheng, P.Y.; Wang, M.; Morgan, E.T. Rapid Transcriptional Suppression of Rat Cytochrome P450 Genes by Endotoxin Treatment and Its Inhibition by Curcumin. J. Pharmacol. Exp. Ther. 2003, 307, 1205–1212. [Google Scholar] [CrossRef]
- Li, X.; Salisbury-Rowswell, J.; Murdock, A.D.; Forse, R.A.; Burke, P.A. Hepatocyte nuclear factor 4 response to injury involves a rapid decrease in DNA binding and transactivation via a JAK2 signal transduction pathway. Biochem. J. 2002, 368, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Ke, S.; Liu, D.; Sheng, T.; Thomas, P.E.; Rabson, A.B.; Gallo, M.A.; Xie, W.; Tian, Y. Role of NF-κB in regulation of PXR-mediated gene expression: A mechanism for the suppression of cytochrome P-450 3A4 by proinflammatory agents. J. Biol. Chem. 2006, 281, 17882–17889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Ke, S.; Denison, M.S.; Rabson, A.B.; Gallo, M.A. Ah receptor and NF-κB interactions, a potential mechanism for dioxin toxicity. J. Biol. Chem. 1999, 274, 510–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, S.; Rabson, A.B.; Germino, J.F.; Gallo, M.A.; Tian, Y. Mechanism of Suppression of Cytochrome P-450 1A1 Expression by Tumor Necrosis Factor-α and Lipopolysaccharide. J. Biol. Chem. 2001, 276, 39638–39644. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Staudinger, J.L. Repression of PXR-mediated induction of hepatic CYP3A gene expression by protein kinase C. Biochem. Pharmacol. 2005, 69, 867–873. [Google Scholar] [CrossRef]
- Lichti-Kaiser, K.; Xu, C.; Staudinger, J.L. Cyclic AMP-dependent protein kinase signaling modulates Pregnane x receptor activity in a species-specific manner. J. Biol. Chem. 2009, 284, 6639–6649. [Google Scholar] [CrossRef] [Green Version]
- Taneja, G.; Chu, C.; Maturu, P.; Moorthy, B.; Ghose, R. Role of c-Jun-N-terminal kinase in pregnane X receptor-mediated induction of human cytochrome P4503A4 in vitro. Drug Metab. Dispos. 2018, 46, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Zimmerman, T.L.; Thevananther, S.; Lee, H.Y.; Kurie, J.M.; Karpen, S.J. Interleukin-1β-mediated suppression of RXR:RAR transactivation of the Ntcp promoter is JNK-dependent. J. Biol. Chem. 2002, 277, 31416–31422. [Google Scholar] [CrossRef] [Green Version]
- Shah, P.; Omoluabi, O.; Moorthy, B.; Ghose, R. Role of adaptor protein toll-like interleukin domain containing adaptor inducing interferon b in toll-like receptor 3- and 4-mediated regulation of hepatic drug metabolizing enzyme and transporter genes. Drug Metab. Dispos. 2016, 44, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Ghose, R.; Zimmerman, T.L.; Thevananther, S.; Karpen, S.J. Endotoxin leads to rapid subcellular re-localization of hepatic RXRα: A novel mechanism for reduced hepatic gene expression in inflammation. Nucl. Recept. 2004, 2. [Google Scholar] [CrossRef] [Green Version]
- Koike, C.; Moore, R.; Negishi, M. Extracellular signal-regulated kinase is an endogenous signal retaining the nuclear Constitutive Active/androstane Receptor (CAR) in the cytoplasm of mouse primary hepatocytes. Mol. Pharmacol. 2007, 71, 1217–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zordoky, B.; El-Kadi, A. Role of NF-kB in the Regulation of Cytochrome P450 Enzymes. Curr. Drug Metab. 2009, 10, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Iber, H.; Chen, Q.; Cheng, P.Y.; Morgan, E.T. Suppression of CYP2C11 gene transcription by interleukin-1 mediated by NF-κB binding at the transcription start site. Arch. Biochem. Biophys. 2000, 377, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Ruminy, P.; Gangneux, C.; Claeyssens, S.; Scotte, M.; Daveau, M.; Salier, J.P. Gene transcription in hepatocytes during the acute phase of a systemic inflammation: From transcription factors to target genes. Inflamm. Res. 2001, 50, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Jover, R.; Bort, R.; Gómez-Lechón, M.J.; Castell, J.V. Down-regulation of human CYP3A4 by the inflammatory signal interleukin-6: Molecular mechanism and transcription factors involved. FASEB J. 2002, 16, 1799–1801. [Google Scholar] [CrossRef]
- Martínez-Jiménez, C.P.; Gómez-Lechón, M.J.; Castell, J.V.; Jover, R. Transcriptional regulation of the human hepatic CYP3A4: Identification of a new distal enhancer region responsive to CCAAT/enhancer-binding protein β isoforms (liver activating protein and liver inhibitory protein). Mol. Pharmacol. 2005, 67, 2088–2101. [Google Scholar] [CrossRef] [Green Version]
- Pitarque, M.; Rodríguez-Antona, C.; Oscarson, M.; Ingelman-Sundberg, M. Transcriptional regulation of the human CYP2A6 gene. J. Pharmacol. Exp. Ther. 2005, 313, 814–822. [Google Scholar] [CrossRef] [Green Version]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef]
- Li, D.; Tolleson, W.H.; Yu, D.; Chen, S.; Guo, L.; Xiao, W.; Tong, W.; Ning, B. MicroRNA-Dependent Gene Regulation of the Human Cytochrome P450. In Pharmacoepigenetics; Elsevier: Amsterdam, The Netherlands, 2019; pp. 129–138. [Google Scholar]
- Papageorgiou, I.; Court, M.H. Identification and validation of microRNAs directly regulating the UDP-glucuronosyltransferase 1A subfamily enzymes by a functional genomics approach. Biochem. Pharmacol. 2017, 137, 93–106. [Google Scholar] [CrossRef]
- Kugler, N.; Klein, K.; Zanger, U.M. MiR-155 and other microRNAs downregulate drug metabolizing cytochromes P450 in inflammation. Biochem. Pharmacol. 2020, 171. [Google Scholar] [CrossRef]
- Morgan, E.T.; Skubic, C.; Lee, C.M.; Cokan, K.B.; Rozman, D. Regulation of cytochrome P450 enzyme activity and expression by nitric oxide in the context of inflammatory disease. Drug Metab. Rev. 2020, 52, 455–471. [Google Scholar] [CrossRef] [PubMed]
- Rae, J.M.; Johnson, M.D.; Lippman, M.E.; Flockhart, D.A. Rifampin is a selective, pleiotropic inducer of drug metabolism genes in human hepatocytes: Studies with cDNA and oligonucleotide expression arrays. J. Pharmacol. Exp. Ther. 2001, 299, 849–857. [Google Scholar] [PubMed]
- Eichelbaum, M.; Mineshita, S.; Ohnhaus, E.; Zekorn, C. The influence of enzyme induction on polymorphic sparteine oxidation. Br. J. Clin. Pharmacol. 1986, 22, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Madan, A.; Graham, R.A.; Carroll, K.M.; Mudra, D.R.; Alayne Burton, L.; Krueger, L.A.; Downey, A.D.; Czerwinski, M.; Forster, J.; Ribadeneira, M.D.; et al. Effects of prototypical microsomal enzyme inducers on cytochrome P450 expression in cultured human hepatocytes. Drug Metab. Dispos. 2003, 31, 421–431. [Google Scholar] [CrossRef] [Green Version]
- Hill, A.V.S. Evolution, revolution and heresy in the genetics of infectious disease susceptibility. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 840–849. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Xu, J.; Fan, J.; Zhang, T.; Li, Y.; Xie, B.; Zhang, W.; Lin, S.; Ye, L.; Liu, Y.; et al. Influence of IL-18 and IL-10 Polymorphisms on Tacrolimus Elimination in Chinese Lung Transplant Patients. Dis. Markers 2017, 2017. [Google Scholar] [CrossRef]
- Xing, J.; Zhang, X.; Fan, J.; Shen, B.; Men, T.; Wang, J. Association between interleukin-18 promoter variants and tacrolimus pharmacokinetics in Chinese renal transplant patients. Eur. J. Clin. Pharmacol. 2015, 71, 191–198. [Google Scholar] [CrossRef]
- Li, Y.; Zou, Y.; Cai, B.; Yang, B.; Ying, B.; Shi, Y.; Wang, L. The associations of IL-18 serum levels and promoter polymorphism with tacrolimus pharmacokinetics and hepatic allograft dysfunction in Chinese liver transplantation recipients. Gene 2012, 491, 251–255. [Google Scholar] [CrossRef]
- Dar, W.A.; Sullivan, E.; Bynon, J.S.; Eltzschig, H.; Ju, C. Ischaemia reperfusion injury in liver transplantation: Cellular and molecular mechanisms. Liver Int. 2019, 39, 788–801. [Google Scholar] [CrossRef] [Green Version]
- Ou, B.; Liu, Y.; Zhang, T.; Sun, Y.; Chen, J.; Peng, Z. TLR9 rs352139 Genetic Variant Promotes Tacrolimus Elimination in Chinese Liver Transplant Patients During the Early Posttransplantation Period. Pharmacotherapy 2019, 39, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wu, S.; Chen, D.; Guo, F.; Zhong, L.; Fan, J.; Peng, Z. Influence of TLR4 rs1927907 locus polymorphisms on tacrolimus pharmacokinetics in the early stage after liver transplantation. Eur. J. Clin. Pharmacol. 2014, 70, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karban, A.S.; Okazaki, T.; Panhuysen, C.I.M.; Gallegos, T.; Potter, J.J.; Bailey-Wilson, J.E.; Silverberg, M.S.; Duerr, R.H.; Cho, J.H.; Gregersen, P.K.; et al. Functional annotation of a novel NFKB1 promoter polymorphism that increases risk for ulcerative colitis. Hum. Mol. Genet. 2004, 13, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, J.L.; Fu, Q.; Wang, X.D.; Liu, L.S.; Wang, C.X.; Xie, W.; Chen, Z.J.; Shu, W.Y.; Huang, M. Associations of ABCB1, NFKB1, CYP3A, and NR1I2 polymorphisms with cyclosporine trough concentrations in Chinese renal transplant recipients. Acta Pharmacol. Sin. 2013, 34, 555–560. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Liu, M.; Wu, X.; Li, G.; Qiu, F.; Gu, J.; Zhao, L. Effect of polymorphisms in CYP3A4, PPARA, NR1I2, NFKB1, ABCG2 and SLCO1B1 on the pharmacokinetics of lovastatin in healthy Chinese volunteers. Pharmacogenomics 2017, 18, 65–75. [Google Scholar] [CrossRef]
- Mbatchi, L.C.; Brouillet, J.P.; Evrard, A. Genetic variations of the xenoreceptors NR1I2 and NR1I3 and their effect on drug disposition and response variability. Pharmacogenomics 2018, 19, 61–77. [Google Scholar] [CrossRef]
- Mulero, M.C.; Wang, V.Y.F.; Huxford, T.; Ghosh, G. Genome reading by the NF-κB transcription factors. Nucleic Acids Res. 2019, 47, 9967–9989. [Google Scholar] [CrossRef] [Green Version]
- Farré, D.; Roset, R.; Huerta, M.; Adsuara, J.E.; Roselló, L.; Albà, M.M.; Messeguer, X. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 2003, 31, 3651–3653. [Google Scholar] [CrossRef] [Green Version]
- Messeguer, X.; Escudero, R.; Farré, D.; Núñez, O.; Martínez, J.; Albà, M.M. PROMO: Detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 2002, 18, 333–334. [Google Scholar] [CrossRef]
- Yevshin, I.; Sharipov, R.; Kolmykov, S.; Kondrakhin, Y.; Kolpakov, F. GTRD: A database on gene transcription regulation—2019 update. Nucleic Acids Res. 2019, 47, D100–D105. [Google Scholar] [CrossRef] [Green Version]
- Dring, M.M.; Goulding, C.A.; Trimble, V.I.; Keegan, D.; Ryan, A.W.; Brophy, K.M.; Smyth, C.M.; Keeling, P.W.N.; O’Donoghue, D.; O’Sullivan, M.; et al. The Pregnane X Receptor Locus Is Associated with Susceptibility to Inflammatory Bowel Disease. Gastroenterology 2006, 130, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Slaviero, K.A.; Clarke, S.J.; Rivory, L.P. Inflammatory response: An unrecognised source of variability in the pharmacokinetics and pharmacodynamics of cancer chemotherapy. Lancet Oncol. 2003, 4, 224–232. [Google Scholar] [CrossRef]
- Mbatchi, L.C.; Robert, J.; Ychou, M.; Boyer, J.C.; Del Rio, M.; Gassiot, M.; Thomas, F.; Tubiana, N.; Evrard, A. Effect of Single Nucleotide Polymorphisms in the Xenobiotic-sensing Receptors NR1I2 and NR1I3 on the Pharmacokinetics and Toxicity of Irinotecan in Colorectal Cancer Patients. Clin. Pharmacokinet. 2016, 55, 1145–1157. [Google Scholar] [CrossRef] [PubMed]
- Klomp, S.D.; Manson, M.L.; Guchelaar, H.-J.; Swen, J.J. Phenoconversion of Cytochrome P450 Metabolism: A Systematic Review. J. Clin. Med. 2020, 9, 2890. [Google Scholar] [CrossRef]
- Veringa, A.; ter Avest, M.; Span, L.F.R.; van den Heuvel, E.R.; Touw, D.J.; Zijlstra, J.G.; Kosterink, J.G.W.; van der Werf, T.S.; Alffenaar, J.W.C. Voriconazole metabolism is influenced by severe inflammation: A prospective study. J. Antimicrob. Chemother. 2017, 72, 261–267. [Google Scholar] [CrossRef]
- Ohnishi, A.; Murakami, S.; Akizuki, S.; Mochizuki, J.; Echizen, H.; Takagi, I. In vivo metabolic activity of CYP2C19 and CYP3A in relation to CYP2C19 genetic polymorphism in chronic liver disease. J. Clin. Pharmacol. 2005, 45, 1221–1229. [Google Scholar] [CrossRef]


{kind=link}
| Stimulus | Model | Effect on CYP mRNA Expression | Effect on Drug Metabolism | Ref. | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Duration | Studied Concentration | Maximal Effect (%) | Potency (EC50 ng/mL) | Duration | Drug | Metabolite | Potency (EC50 ng/mL) | Maximal Effect (%) | |||
| IL-6 | PHH | 24 h | 10 ng/mL | CYP3A4 (↓95%) | [14] | ||||||
| CYP2C9 (↓35%) | |||||||||||
| CYP2C19 (↓40%) | |||||||||||
| PHH | 24 h | 10 ng/mL | CYP3A4 (↓85%) | 72 h | atorvastatin | o-OH-atorvastatin | NS | [15] | |||
| CYP1A2 (↓76%) | phenacetin | acetaminophen | NS | ||||||||
| CYP2C9 (↓65%) | tolbutamide | OH-tolbutamide | NS | ||||||||
| CYP2C19 (↓41%) | S-mephenytoin | 4′-OH-mephentoin | NS | ||||||||
| CYP2D6 (↓41%) | |||||||||||
| CYP2E1 (↑402%) | |||||||||||
| PHH | 24 h | 10 ng/mL | CYP3A4 (↓97%) | [16] | |||||||
| CYP1A2 (↓96%) | |||||||||||
| PHH | 48 h | 0.0006–50 ng/mL | CYP3A4 (↓90%) | 0.454 | [17] | ||||||
| CYP1A2 (↓80%) | 5.49 | ||||||||||
| PHH * | 48 h | 10 ng/mL | CYP3A4 (↓98%) | 72 h | testosterone | 6β-hydroxytestosterone | ↓76% | [18] | |||
| CYP1A2 (↓27%) | phenacetin | acetaminophen | ↓22% | ||||||||
| CYP2C19 (↓72%) | S-mephenytoin | 4′-OH-mephentoin | ↓65% | ||||||||
| CYP2C9 (↓63%) | tolbutamide | OH-tolbutamide | ↓35% | ||||||||
| CYP2D6 (↑240%) | dextromethorphan | dextrorphan | ↓39% | ||||||||
| PHH | 72 h | 0.005–50 ng/mL | CYP3A4 (↓95%) | 0.0032 | 72 h | testosterone | 6β-hydroxytestosterone | 0.073 | ↓70% | [19] | |
| CYP3A5 (↓95%) | 0.051 | ||||||||||
| CYP1A2 (↓85%) | 0.271 | phenacetin | acetaminophen | 1.25 | ↓90% | ||||||
| CYP2C19 (↓80%) | 0.071 | ||||||||||
| CYP2C9 (↓90%) | 0.121 | ||||||||||
| CYP2D6 (↓70%) | 0.151 | ||||||||||
| PHH/PHH:KC (10:4) | 2–200 ng/mL | CYP3A4 (?) | 72 h | testosterone | 6β-hydroxytestosterone | ↓90% | [20] | ||||
| PHH:KC * | 0.001–10 ng/mL | CYP3A4 (?) | 96 h | luminogenic P450-Glo™ substrate | proluciferin substrate | 0.463 | ↓80% | [21] | |||
| PHH:KC (10:4) | 96 h | 0.00625–5 ng/mL | CYP3A4 (↓95%) | 96 h | luminogenic P450-Glo™ substrate | proluciferin substrate | 0.252 | ↓ > 95% | [22] | ||
| HepaRG | 24 h | 10 ng/mL | CYP3A4 (↓95%) | 24 h | midazolam | 1′-hydroxymidazolam | decreased | [23] | |||
| CYP3A5 (↓90%) | |||||||||||
| CYP1A2 (↓80%) | phenacetin | acetaminophen | decreased | ||||||||
| CYP2C9 (↓85%) | tolbutamide | OH-tolbutamide | NS | ||||||||
| CYP2C19 (↓85%) | S-mephenytoin | 4′-OH-mephentoin | decreased | ||||||||
| CYP2D6 (NS) | propafenone | 5-hydroxypropafenone | NS | ||||||||
| CYP2E1 (NS) | |||||||||||
| HepaRG | 24 h | 10 ng/mL | CYP3A4 (↓93%) | 72 h | atorvastatin | o-OH-atorvastatin | ↓ > 80% | [15] | |||
| CYP3A5 (↓89%) | |||||||||||
| CYP1A2 (↓90%) | phenacetin | acetaminophen | ↓ > 60% | ||||||||
| CYP2C9 (↓83%) | tolbutamide | OH-tolbutamide | ↓ > 60% | ||||||||
| CYP2C19 (↓83%) | S-mephenytoin | 4′-OH-mephentoin | ↓ > 60% | ||||||||
| CYP2E1 (NS) | |||||||||||
| HepaRG | 48 h | 0.123–30 ng/mL | CYP3A4 (↓99%) | <0.123 | 72 h | midazolam | 1′-hydroxymidazolam | 2.89 | ↓60% | [17] | |
| CYP1A2 (↓90%) | 0.452 | phenacetin | acetaminophen | 8.96 | ↓65% | ||||||
| HepaRG | 48 h | 10 ng/mL | CYP3A4 (↓ > 95%) | 4 h | midazolam | 1′-hydroxymidazolam | ↓61% | [16] | |||
| CYP1A2 (↓ > 95%) | phenacetin | acetaminophen | ↓68% | ||||||||
| HepaRG | 336 h (14 days) | 10 ng/mL | CYP3A4 (NS) | 336 h (14 days) | midazolam | 1′-hydroxymidazolam | decreased | [23] | |||
| CYP3A5 (↓80%) | |||||||||||
| CYP1A2 (↓95%) | phenacetin | acetaminophen | decreased | ||||||||
| CYP2C9 (NS) | tolbutamide | OH-tolbutamide | NS | ||||||||
| CYP2C19 (↓90%) | S-mephenytoin | 4′-OH-mephentoin | NS | ||||||||
| CYP2D6 (NS) | |||||||||||
| CYP2E1 (NS) | |||||||||||
| IL-1β | PHH | 24 h | 5 ng/mL | CYP3A4 (↓95%) | [14] | ||||||
| CYP2C9 (NS) | |||||||||||
| CYP2C19 (NS) | |||||||||||
| PHH | 72 h | 0.0001–10 ng/mL | CYP3A4 (↓95%) | 0.294 | 72 h | testosterone | 6β-hydroxytestosterone | 0.416 | ↓90% | [24] | |
| CYP3A5 (↓62%) | 0.347 | ||||||||||
| CYP1A2 (↓73%) | 0.531 # | phenacetin | acetaminophen | 0.45 | ↓65% | ||||||
| CYP2C9 (↓79%) | 0.229 # | ||||||||||
| CYP2C19 (↓58%) | 0.153 # | ||||||||||
| CYP2D6 (↓75%) | 0.945 # | ||||||||||
| PHH/ PHH:KC (10:4) | 0.2–200 ng/mL | CYP3A4 (?) | 72 h | Testosterone | 6β-hydroxytestosterone | ↓85% | [20] | ||||
| PHH:KC (10:4) | 96 h | 0.00625–5 ng/mL | CYP3A4 (↓ > 95%) | 96 h | luminogenic P450-Glo™ substrate | proluciferin substrate | 0.098 | ↓ > 95% | [22] | ||
| HepaRG | 24 h | 5 ng/mL | CYP3A4 (↓97%) | 72 h | atorvastatin | o-OH-atorvastatin | ↓ > 80% | [15] | |||
| CYP3A5 (↓91%) | |||||||||||
| CYP1A2 (↓93%) | phenacetin | acetaminophen | ↓ > 80% | ||||||||
| CYP2C9 (↓90%) | tolbutamide | OH-tolbutamide | ↓ > 80% | ||||||||
| CYP2C19 (↓93%) | S-mephenytoin | 4′-OH-mephentoin | ↓ > 80% | ||||||||
| CYP2E1 (↓75%) | |||||||||||
| HepaRG | 24 h | 1 ng/mL | CYP3A4 (↓98%) | [16] | |||||||
| CYP1A2 (↓99%) | |||||||||||
| IL-18 | PHH | 48 h | 1.95–500 ng/mL | CYP3A4 (NS) | 72 h | midazolam | 1′-hydroxymidazolam | NS | [17] | ||
| CYP1A2 (NS) | phenacetin | acetaminophen | NS | ||||||||
| HepaRG | 48 h | 2.06–600 ng/mL | CYP3A4 (NS) | 72 h | midazolam | 1′-hydroxymidazolam | NS | [17] | |||
| CYP1A2 (NS) | phenacetin | acetaminophen | NS | ||||||||
| TNF-α | PHH | 24 h | 10 ng/mL | CYP3A4 (↓80%) | [14] | ||||||
| CYP2C9 (NS) | |||||||||||
| CYP2C19 (NS) | |||||||||||
| PHH * | 48 h | 10 ng/mL | CYP3A4 (↓87%) | 72 h | testosterone | 6β-hydroxytestosterone | ↓70% | [18] | |||
| CYP1A2 (↓45%) | phenacetin | acetaminophen | ↓72% | ||||||||
| CYP2C19 (NS) | S-mephenytoin | 4′-OH-mephentoin | ↓82% | ||||||||
| CYP2C9 (NS) | tolbutamide | OH-tolbutamide | ↓17% | ||||||||
| CYP2D6 (↓40%) | dextromethorphan | dextrorphan | ↓42% | ||||||||
| HepaRG | 24 h | 10 ng/mL | CYP3A4 (↓90%) | 72 h | atorvastatin | o-OH-atorvastatin | ↓ > 80% | [15] | |||
| CYP3A5 (↓79%) | |||||||||||
| CYP1A2 (↓87%) | phenacetin | acetaminophen | ↓ > 80% | ||||||||
| CYP2C19 (↓64%) | S-mephenytoin | 4′-OH-mephentoin | ↓ > 80% | ||||||||
| CYP2C9 (↓62%) | tolbutamide | OH-tolbutamide | ↓ > 80% | ||||||||
| CYP2E1 (↓54%) | |||||||||||
| TGF-β | PHH | 24 h | 10 ng/mL | CYP3A4 (↓75%) | [14] | ||||||
| CYP2C9 (↓50%) | |||||||||||
| CYP2C19 (↓50%) | |||||||||||
| IFN-y | PHH | 24 h | 10 ng/mL | CYP3A4 (↓75%) | [14] | ||||||
| CYP2C9 (NS) | |||||||||||
| CYP2C19 (NS) | |||||||||||
| IL-22 | PHH | 48 h | 10 ng/mL | CYP3A4 (↓70%) | [25] | ||||||
| CYP1A2 (↓45%) | |||||||||||
| CYP2C9 (↓50%) | |||||||||||
| HepaRG | 24 h | 10 ng/mL | CYP3A4 (↓75%) | 1.7 | 48 h | midazolam | 1′-hydroxymidazolam | ↓50% | [25] | ||
| CYP1A2 (↓60%) | phenacetin | acetaminophen | ↓50% | ||||||||
| CYP2C9 (↓50%) | |||||||||||
| IL-2 | PHH * | 2–200 ng/mL | CYP3A4 (?) | 72 h | testosterone | 6β-hydroxytestosterone | NS | [20] | |||
| PHH * | 48 h | 10 ng/mL | CYP3A4 (NS) | 72 h | testosterone | 6β-hydroxytestosterone | NS | [18] | |||
| CYP1A2 (NS) | phenacetin | acetaminophen | NS | ||||||||
| CYP2C19 (NS) | S-mephenytoin | 4′-OH-mephentoin | ↓21% | ||||||||
| CYP2C9 (NS) | tolbutamide | OH-tolbutamide | NS | ||||||||
| CYP2D6 (↑150%) | dextromethorphan | dextrorphan | ↓22% | ||||||||
| PHH:KC (10:4) * | 200 ng/mL | CYP3A4 (?) | 72 h | testosterone | 6β-hydroxytestosterone | ↓70% | [20] | ||||
| PHH:KC (10:4) | 200 ng/mL | CYP3A4 (?) | 96 h | luminogenic P450-Glo™ substrate | proluciferin substrate | NS | [22] | ||||
| IL-12 | PHH | 48 h | 10 ng/mL | CYP3A4 (NS) | 48 h | testosterone | 6β-hydroxytestosterone | NS | [26] | ||
| CYP2C19 (NS) | S-mephenytoin | 4′-OH-mephentoin | |||||||||
| CYP2C9 (NS) | tolbutamide | OH-tolbutamide | |||||||||
| IL-23 | PHH | 48 h | 10 ng/mL | CYP3A4 (NS) | 48 h | testosterone | 6β-hydroxytestosterone | NS | [26] | ||
| CYP2C19 (NS) | S-mephenytoin | 4′-OH-mephentoin | |||||||||
| CYP2C9 (NS) | tolbutamide | OH-tolbutamide | |||||||||
| PHH:KC (10:4) | 200 ng/mL | CYP3A4 (?) | 96 h | luminogenic P450-Glo™ substrate | proluciferin substrate | NS | [22] | ||||
| LPS | PHH | 24 h | 10 µg/ml | CYP3A4 (↓95%) | [14] | ||||||
| CYP2C9 (NS) | |||||||||||
| CYP2C19 (NS) | |||||||||||
| HepaRG | 48 h | 1.37–333 ng/mL | CYP3A4 (↓95%) | <1.37 | 72 h | midazolam | 1′-hydroxymidazolam | 7.85 | ↓60% | [17] | |
| SNP | Variation | Location | Allele Frequency | In Binding Site (Proximity) of: | Distance to Binding Site (bp) | Binding Spot Predicted in: |
|---|---|---|---|---|---|---|
| rs10934498 | G > A, C, T | intron | G = 0.5024 | NFκB1-p105 subunit | 0 | GTRD |
| A = 0.4976 | ||||||
| rs1403526 | A > C, G | Intron | A = 0.64900 | RelA-p65 subunit | 0 | Alggen PROMO |
| G = 0.35100 | ||||||
| rs12721602 | G > A | 5 -UTR | G = 0.98303 | RelA-p65 subunit | 13 | Alggen PROMO |
| A = 0.01697 | ||||||
| rs1054191 | G > A, C | 3′-UTR | G = 0.87745 | NF-κB, NF-κB1 p105 | 17 | Alggen PROMO |
| A = 0.12255 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Jong, L.M.; Jiskoot, W.; Swen, J.J.; Manson, M.L. Distinct Effects of Inflammation on Cytochrome P450 Regulation and Drug Metabolism: Lessons from Experimental Models and a Potential Role for Pharmacogenetics. Genes 2020, 11, 1509. https://doi.org/10.3390/genes11121509
de Jong LM, Jiskoot W, Swen JJ, Manson ML. Distinct Effects of Inflammation on Cytochrome P450 Regulation and Drug Metabolism: Lessons from Experimental Models and a Potential Role for Pharmacogenetics. Genes. 2020; 11(12):1509. https://doi.org/10.3390/genes11121509
Chicago/Turabian Stylede Jong, Laura M., Wim Jiskoot, Jesse J. Swen, and Martijn L. Manson. 2020. "Distinct Effects of Inflammation on Cytochrome P450 Regulation and Drug Metabolism: Lessons from Experimental Models and a Potential Role for Pharmacogenetics" Genes 11, no. 12: 1509. https://doi.org/10.3390/genes11121509
APA Stylede Jong, L. M., Jiskoot, W., Swen, J. J., & Manson, M. L. (2020). Distinct Effects of Inflammation on Cytochrome P450 Regulation and Drug Metabolism: Lessons from Experimental Models and a Potential Role for Pharmacogenetics. Genes, 11(12), 1509. https://doi.org/10.3390/genes11121509