Technologies for Pharmacogenomics: A Review



Abstract
:1. Introduction
2. SNV Panels: Current Clinical Practice
2.1. Commercial Arrays
2.2. Custom Arrays
2.3. Array Developments
3. Next Generation Sequencing
3.1. Next Generation Sequencing Technologies
3.2. Use of NGS for Pharmacogenomics
3.3. Repurposing of Clinical Genetics Data
4. Long-Read Sequencing
4.1. Long-Read Sequencing
4.2. Long-Read Sequencing for PGx
5. Challenges
5.1. Drug Metabolizer Phenotype Inference
5.2. Imputation
5.3. Haplotype Phasing
5.4. Structural Variants
5.5. Variants of Unknown Effect
5.6. Pharmacogenomics and Disease Genes
Author Contributions
Funding
Conflicts of Interest
References
- Kirchheiner, J.; Brøsen, K.; Dahl, M.L.; Gram, L.F.; Kasper, S.; Roots, I.; Sjöqvist, F.; Spina, E.; Brockmöller, J. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: A first step towards subpopulation-specific dosages. Acta Psychiatr. Scand. 2001, 104, 173–192. [Google Scholar] [CrossRef] [PubMed]
- Swen, J.J.; Wilting, I.; de Goede, A.L.; Grandia, L.; Mulder, H.; Touw, D.J.; de Boer, A.; Conemans, J.M.; Egberts, T.C.; Klungel, O.H.; et al. Pharmacogenetics: From bench to byte. Clin. Pharmacol. Ther. 2008, 83, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.M.; Haidar, C.E.; Wilkinson, M.R.; Crews, K.R.; Baker, D.K.; Kornegay, N.M.; Yang, W.; Pui, C.H.; Reiss, U.M.; Gaur, A.H.; et al. PG4KDS: A model for the clinical implementation of pre-emptive pharmacogenetics. Am. J. Med. Genet. C Semin. Med. Genet. 2014, 166c, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Dutch Pharmacogenetics Working group. Pharmacogenetics Guidelines; Royal Dutch Pharmacists Association (KNMP Kennisbank): The Hague, The Netherlands, 2020. [Google Scholar]
- Clinical Pharmacogenetics Implementation Consortium. CPIC-guidelines. Available online: https://cpicpgx.org/ (accessed on 16 October 2020).
- National Human Genome Research Institute. DNA Sequencing Costs: Data. Available online: https://www.genome.gov/about-genomics/fact-sheets/DNA-Sequencing-Costs-Data (accessed on 16 October 2020).
- Hallberg, P.; Yue, Q.Y.; Eliasson, E.; Melhus, H.; Ås, J.; Wadelius, M. SWEDEGENE-a Swedish nation-wide DNA sample collection for pharmacogenomic studies of serious adverse drug reactions. Pharm. J. 2020, 20, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Reisberg, S.; Krebs, K.; Kals, M.; Magi, R.; Metsalu, K.; Lauschke, V.M.; Vilo, J.; Milani, L. Translating genotype data of 44,000 biobank participants into clinical pharmacogenetic recommendations: Challenges and solutions. Genet. Med. 2019, 21, 1345–1354. [Google Scholar] [CrossRef] [Green Version]
- Cousin, M.A.; Matey, E.T.; Blackburn, P.R.; Boczek, N.J.; McAllister, T.M.; Kruisselbrink, T.M.; Babovic-Vuksanovic, D.; Lazaridis, K.N.; Klee, E.W. Pharmacogenomic findings from clinical whole exome sequencing of diagnostic odyssey patients. Mol. Genet. Genom. Med. 2017, 5, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Van der Lee, M.; Allard, W.G.; Bollen, S.; Santen, G.W.E.; Ruivenkamp, C.A.L.; Hoffer, M.J.V.; Kriek, M.; Guchelaar, H.J.; Anvar, S.Y.; Swen, J.J. Repurposing of Diagnostic Whole Exome Sequencing Data of 1,583 Individuals for Clinical Pharmacogenetics. Clin. Pharmacol. Ther. 2020, 107, 617–627. [Google Scholar] [CrossRef] [Green Version]
- Krebs, K.; Milani, L. Translating pharmacogenomics into clinical decisions: Do not let the perfect be the enemy of the good. Hum. Genom. 2019, 13, 39. [Google Scholar] [CrossRef] [Green Version]
- Ameur, A.; Kloosterman, W.P.; Hestand, M.S. Single-molecule sequencing: Towards clinical applications. Trends Biotechnol. 2019, 37, 72–85. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Sereewattanawoot, S.; Suzuki, A. A new era of long-read sequencing for cancer genomics. J Hum. Genet. 2020, 65, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Aganezov, S.; Goodwin, S.; Sherman, R.M.; Sedlazeck, F.J.; Arun, G.; Bhatia, S.; Lee, I.; Kirsche, M.; Wappel, R.; Kramer, M.; et al. Comprehensive analysis of structural variants in breast cancer genomes using single-molecule sequencing. Genome Res. 2020, 30, 1258–1273. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Xu, L.; Seki, M.; Yokoyama, T.T.; Kasahara, M.; Kashima, Y.; Ohashi, A.; Shimada, Y.; Motoi, N.; Tsuchihara, K.; et al. Long-read sequencing for non-small-cell lung cancer genomes. Genome Res. 2020, 30, 1243–1257. [Google Scholar] [CrossRef] [PubMed]
- Just, K.S.; Steffens, M.; Swen, J.J.; Patrinos, G.P.; Guchelaar, H.J.; Stingl, J.C. Medical education in pharmacogenomics-results from a survey on pharmacogenetic knowledge in healthcare professionals within the European pharmacogenomics clinical implementation project Ubiquitous Pharmacogenomics (U-PGx). Eur. J. Clin. Pharmacol. 2017, 73, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Rollinson, V.; Turner, R.; Pirmohamed, M. Pharmacogenomics for Primary Care: An Overview. Genes 2020, 11, 1337. [Google Scholar] [CrossRef]
- Bank, P.C.D.; Swen, J.J.; Guchelaar, H.J. Implementation of Pharmacogenomics in Everyday Clinical Settings. Stud. Surf. Sci. Catal. 2018, 83, 219–246. [Google Scholar] [CrossRef]
- PharmGKB. DPWG: Dutch Pharmacogenetics Working Group. Available online: https://www.pharmgkb.org/page/dpwg (accessed on 23 June 2020).
- Illumina Inc. VeraCode ADME Core Panel. Available online: https://www.illumina.com/documents/products/datasheets/datasheet_veracode_adme_core_panel.pdf (accessed on 21 July 2020).
- Arbitrio, M.; Di Martino, M.T.; Scionti, F.; Agapito, G.; Guzzi, P.H.; Cannataro, M.; Tassone, P.; Tagliaferri, P. DMET™ (Drug Metabolism Enzymes and Transporters): A pharmacogenomic platform for precision medicine. Oncotarget 2016, 7, 54028–54050. [Google Scholar] [CrossRef]
- ThermoFisher Scientific. Pharmacoscan Assay. Available online: https://www.thermofisher.com/order/catalog/product/903010TS (accessed on 16 October 2020).
- Gabriel, S.; Ziaugra, L.; Tabbaa, D. SNP genotyping using the Sequenom MassARRAY iPLEX platform. Curr. Protoc. Hum. Genet. 2009. [Google Scholar] [CrossRef]
- Spierings, G.; Dunbar, S.A. Pharmacogenetics using Luminex(R) xMAP(R) technology: A method for developing a custom multiplex single nucleotide polymorphism mutation assay. Methods Mol. Biol. 2013, 1015, 115–126. [Google Scholar] [CrossRef]
- Chen, C.; Li, S.; Lu, X.; Tan, B.; Huang, C.; Qin, L. High resolution melting method to detect single nucleotide polymorphism of VKORC1 and CYP2C9. Int. J. Clin. Exp. Pathol. 2014, 7, 2558–2564. [Google Scholar]
- Jannetto, P.J.; Laleli-Sahin, E.; Wong, S.H. Pharmacogenomic genotyping methodologies. Clin. Chem. Lab. Med. 2004, 42, 1256–1264. [Google Scholar] [CrossRef]
- Ghasemi, Z.; Hashemi, M.; Ejabati, M.; Ebrahimi, S.M.; Kheiri Manjili, H.; Sharafi, A.; Ramazani, A. Development of a High-Resolution Melting Analysis Method for CYP2C19*17 Genotyping in Healthy Volunteers. Avicenna J. Med. Biotechnol. 2016, 8, 193–199. [Google Scholar] [PubMed]
- Mukerjee, G.; Huston, A.; Kabakchiev, B.; Piquette-Miller, M.; van Schaik, R.; Dorfman, R. User considerations in assessing pharmacogenomic tests and their clinical support tools. NPJ Genom. Med. 2018, 3, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilella, A. Next-Generation-Sequencing, v1.5.7. Available online: https://docs.google.com/spreadsheets/d/1GMMfhyLK0-q8XkIo3YxlWaZA5vVMuhU1kg41g4xLkXc/htmlview?hl=en_GB (accessed on 27 October 2020).
- Agenda Bioscience. VeriDose Core Panel. Available online: https://agenabio.com/products/panel/veridose-core-panel/ (accessed on 2 November 2020).
- Huang, S.M.; Goodsaid, F.; Rahman, A.; Frueh, F.; Lesko, L.J. Application of pharmacogenomics in clinical pharmacology. Toxicol. Mech. Methods. 2006, 16, 89–99. [Google Scholar] [CrossRef] [PubMed]
- ThermoFisher Scientifuic. Axiom Pharmacofocus. Available online: https://www.thermofisher.com/order/catalog/product/952425?SID=srch-hj-952425#/952425?SID=srch-hj-952425 (accessed on 3 November 2020).
- Pulley, J.M.; Denny, J.C.; Peterson, J.F.; Bernard, G.R.; Vnencak-Jones, C.L.; Ramirez, A.H.; Delaney, J.T.; Bowton, E.; Brothers, K.; Johnson, K.; et al. Operational implementation of prospective genotyping for personalized medicine: The design of the Vanderbilt PREDICT project. Clin. Pharmacol. Ther. 2012, 92, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Van der Wouden, C.H.; Cambon-Thomsen, A.; Cecchin, E.; Cheung, K.C.; Davila-Fajardo, C.L.; Deneer, V.H.; Dolzan, V.; Ingelman-Sundberg, M.; Jonsson, S.; Karlsson, M.O.; et al. Implementing Pharmacogenomics in Europe: Design and Implementation Strategy of the Ubiquitous Pharmacogenomics Consortium. Clin. Pharmacol. Ther. 2017, 101, 341–358. [Google Scholar] [CrossRef]
- ThermoFisher Scientific. Open Array Technology Overview. Available online: https://www.thermofisher.com/nl/en/home/life-science/pcr/real-time-pcr/real-time-openarray/open-array-technology.html (accessed on 3 November 2020).
- Eadon, M.T.; Desta, Z.; Levy, K.D.; Decker, B.S.; Pierson, R.C.; Pratt, V.M.; Callaghan, J.T.; Rosenman, M.B.; Carpenter, J.S.; Holmes, A.M.; et al. Implementation of a pharmacogenomics consult service to support the INGENIOUS trial. Clin. Pharmacol. Ther. 2016, 100, 63–66. [Google Scholar] [CrossRef]
- Van der Wouden, C.H.; van Rhenen, M.H.; Jama, W.O.M.; Ingelman-Sundberg, M.; Lauschke, V.M.; Konta, L.; Schwab, M.; Swen, J.J.; Guchelaar, H.J. Development of the PGx-Passport: A Panel of Actionable Germline Genetic Variants for Pre-emptive Pharmacogenetic Testing. Clin. Pharmacol. Ther. 2019, 106, 866–873. [Google Scholar] [CrossRef]
- Biosearch Technologies. SNPline Genotyping Automation. Available online: https://www.biosearchtech.com/products/instruments-and-consumables/genotyping-instruments/snpline-genotyping-automation (accessed on 3 November 2020).
- Illumina Inc. Illumina Global Screening Array. Available online: https://emea.illumina.com/products/by-type/microarray-kits/infinium-global-screening.html (accessed on 23 June 2020).
- ThermoFisher Scientific. Axiom Genotyping Solutions. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/brochures/axiom_solution_brochure.pdf (accessed on 3 November 2020).
- Ingelman-Sundberg, M. Pharmacogenetics of cytochrome P450 and its applications in drug therapy: The past, present and future. Trends Pharmacol. Sci. 2004, 25, 193–200. [Google Scholar] [CrossRef]
- Gaedigk, A.; Ingelman-Sundberg, M.; Miller, N.A.; Leeder, J.S.; Whirl-Carrillo, M.; Klein, T.E. The Pharmacogene Variation (PharmVar) Consortium: Incorporation of the Human Cytochrome P450 (CYP) Allele Nomenclature Database. Clin. Pharmacol. Ther. 2018, 103, 399–401. [Google Scholar] [CrossRef] [Green Version]
- Broadinstitute. GnomAD. Available online: https://gnomad.broadinstitute.org/ (accessed on 26 October 2020).
- Mardis, E.R. Next-generation sequencing platforms. Annu. Rev. Anal. Chem. 2013, 6, 287–303. [Google Scholar] [CrossRef] [Green Version]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.E.; Myers, R.M. Advancements in Next-Generation Sequencing. Annu. Rev. Genom. Hum. Genet. 2016, 17, 95–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Wu, G.; Broeckel, U.; Smith, C.A.; Turner, V.; Haidar, C.E.; Wang, S.; Carter, R.; Karol, S.E.; Neale, G.; et al. Comparison of genome sequencing and clinical genotyping for pharmacogenes. Clin. Pharmacol. Ther. 2016, 100, 380–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Londin, E.R.; Clark, P.; Sponziello, M.; Kricka, L.J.; Fortina, P.; Park, J.Y. Performance of exome sequencing for pharmacogenomics. Pers. Med. 2014, 12, 109–115. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen-Torvik, L.J.; Almoguera, B.; Doheny, K.F.; Freimuth, R.R.; Gordon, A.S.; Hakonarson, H.; Hawkins, J.B.; Husami, A.; Ivacic, L.C.; Kullo, I.J.; et al. Concordance between Research Sequencing and Clinical Pharmacogenetic Genotyping in the eMERGE-PGx Study. J. Mol. Diagn. 2017, 19, 561–566. [Google Scholar] [CrossRef] [Green Version]
- Ng, D.; Hong, C.S.; Singh, L.N.; Johnston, J.J.; Mullikin, J.C.; Biesecker, L.G. Assessing the capability of massively parallel sequencing for opportunistic pharmacogenetic screening. Genet. Med. 2017, 19, 357–361. [Google Scholar] [CrossRef] [Green Version]
- Cohn, I.; Paton, T.A.; Marshall, C.R.; Basran, R.; Stavropoulos, D.J.; Ray, P.N.; Monfared, N.; Hayeems, R.Z.; Meyn, M.S.; Bowdin, S.; et al. Genome sequencing as a platform for pharmacogenetic genotyping: A pediatric cohort study. NPJ Genom. Med. 2017, 2, 19. [Google Scholar] [CrossRef]
- Gordon, A.S.; Fulton, R.S.; Qin, X.; Mardis, E.R.; Nickerson, D.A.; Scherer, S. PGRNseq: A targeted capture sequencing panel for pharmacogenetic research and implementation. Pharm. Genom. 2016, 26, 161–168. [Google Scholar] [CrossRef]
- Chua, E.W.; Cree, S.L.; Ton, K.N.; Lehnert, K.; Shepherd, P.; Helsby, N.; Kennedy, M.A. Cross-Comparison of Exome Analysis, Next-Generation Sequencing of Amplicons, and the iPLEX((R)) ADME PGx Panel for Pharmacogenomic Profiling. Front. Pharmacol. 2016, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- Ingelman-Sundberg, M.; Sim, S.C. Intronic polymorphisms of cytochromes P450. Hum. Genom. 2010, 4, 402–405. [Google Scholar] [CrossRef] [Green Version]
- Bush, W.S.; Crosslin, D.R.; Owusu-Obeng, A.; Wallace, J.; Almoguera, B.; Basford, M.A.; Bielinski, S.J.; Carrell, D.S.; Connolly, J.J.; Crawford, D.; et al. Genetic variation among 82 pharmacogenes: The PGRNseq data from the eMERGE network. Pharmacol. Ther. 2016, 100, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Gulilat, M.; Lamb, T.; Teft, W.A.; Wang, J.; Dron, J.S.; Robinson, J.F.; Tirona, R.G.; Hegele, R.A.; Kim, R.B.; Schwarz, U.I. Targeted next generation sequencing as a tool for precision medicine. BMC Med. Genom. 2019, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Aquilante, C.L.; Kao, D.P.; Trinkley, K.E.; Lin, C.T.; Crooks, K.R.; Hearst, E.C.; Hess, S.J.; Kudron, E.L.; Lee, Y.M.; Liko, I.; et al. Clinical implementation of pharmacogenomics via a health system-wide research biobank: The University of Colorado experience. Pharmacogenomics 2020, 21, 375–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Institute of Health. AllofUs Research Program. Available online: https://allofus.nih.gov/ (accessed on 23 October 2020).
- Precision Medicine Initiative Work Group. The Precision Medicine Initiative Cohort Program—Building a Research Foundation for 21st Century Medicine; National Institutes of Health. 2015. Available online: https://www.nih.gov/sites/default/files/research-training/initiatives/pmi/pmi-working-group-report-20150917-2.pdf (accessed on 23 October 2020).
- Caspar, S.M.; Schneider, T.; Meienberg, J.; Matyas, G. Added Value of Clinical Sequencing: WGS-Based Profiling of Pharmacogenes. Int. J. Mol. Sci. 2020, 21, 2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantere, T.; Kersten, S.; Hoischen, A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019, 10, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Bowden, R.; Davies, R.W.; Heger, A.; Pagnamenta, A.T.; de Cesare, M.; Oikkonen, L.E.; Parkes, D.; Freeman, C.; Dhalla, F.; Patel, S.Y.; et al. Sequencing of human genomes with nanopore technology. Nat. Commun. 2019, 10, 1869. [Google Scholar] [CrossRef]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef]
- Schüle, B.; McFarland, K.N.; Lee, K.; Tsai, Y.C.; Nguyen, K.D.; Sun, C.; Liu, M.; Byrne, C.; Gopi, R.; Huang, N.; et al. Parkinson’s disease associated with pure ATXN10 repeat expansion. NPJ Park. Dis. 2017, 3, 27. [Google Scholar] [CrossRef]
- Ardui, S.; Race, V.; Zablotskaya, A.; Hestand, M.S.; Van Esch, H.; Devriendt, K.; Matthijs, G.; Vermeesch, J.R. Detecting AGG Interruptions in Male and Female FMR1 Premutation Carriers by Single-Molecule Sequencing. Hum. Mutat. 2017, 38, 324–331. [Google Scholar] [CrossRef] [Green Version]
- Qiao, W.; Yang, Y.; Sebra, R.; Mendiratta, G.; Gaedigk, A.; Desnick, R.J.; Scott, S.A. Long-Read Single Molecule Real-Time Full Gene Sequencing of Cytochrome P450-2D6. Hum. Mutat. 2016, 37, 315–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buermans, H.P.; Vossen, R.H.; Anvar, S.Y.; Allard, W.G.; Guchelaar, H.J.; White, S.J.; den Dunnen, J.T.; Swen, J.J.; van der Straaten, T. Flexible and Scalable Full-Length CYP2D6 Long Amplicon PacBio Sequencing. Hum. Mutat. 2017, 38, 310–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robarge, J.D.; Li, L.; Desta, Z.; Nguyen, A.; Flockhart, D.A. The star-allele nomenclature: Retooling for translational genomics. Clin. Pharmacol. Ther. 2007, 82, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Numanagic, I.; Malikic, S.; Ford, M.; Qin, X.; Toji, L.; Radovich, M.; Skaar, T.C.; Pratt, V.M.; Berger, B.; Scherer, S.; et al. Allelic decomposition and exact genotyping of highly polymorphic and structurally variant genes. Nat. Commun. 2018, 9, 828. [Google Scholar] [CrossRef] [PubMed]
- Twist, G.P.; Gaedigk, A.; Miller, N.A.; Farrow, E.G.; Willig, L.K.; Dinwiddie, D.L.; Petrikin, J.E.; Soden, S.E.; Herd, S.; Gibson, M.; et al. Constellation: A tool for rapid, automated phenotype assignment of a highly polymorphic pharmacogene, CYP2D6, from whole-genome sequences. NPJ Genom. Med. 2016, 1, 15007. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Wheeler, M.M.; Thummel, K.E.; Nickerson, D.A. Calling Star Alleles with Stargazer in 28 Pharmacogenes With Whole Genome Sequences. Clin. Pharmacol. Ther. 2019, 106, 1328–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twesigomwe, D.; Wright, G.E.B.; Drögemöller, B.I.; da Rocha, J.; Lombard, Z.; Hazelhurst, S. A systematic comparison of pharmacogene star allele calling bioinformatics algorithms: A focus on CYP2D6 genotyping. NPJ Genom. Med. 2020, 5, 30. [Google Scholar] [CrossRef]
- Pratt, V.M.; Everts, R.E.; Aggarwal, P.; Beyer, B.N.; Broeckel, U.; Epstein-Baak, R.; Hujsak, P.; Kornreich, R.; Liao, J.; Lorier, R.; et al. Characterization of 137 Genomic DNA Reference Materials for 28 Pharmacogenetic Genes: A GeT-RM Collaborative Project. J. Mol. Diagn. 2016, 18, 109–123. [Google Scholar] [CrossRef] [Green Version]
- Browning, S.R.; Browning, B.L. Haplotype phasing: Existing methods and new developments. Nat. Rev. Genet. 2011, 12, 703–714. [Google Scholar] [CrossRef] [Green Version]
- McCormack, M.; Alfirevic, A.; Bourgeois, S.; Farrell, J.J.; Kasperavičiūtė, D.; Carrington, M.; Sills, G.J.; Marson, T.; Jia, X.; de Bakker, P.I.; et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N. Engl. J. Med. 2011, 364, 1134–1143. [Google Scholar] [CrossRef] [Green Version]
- Ozeki, T.; Mushiroda, T.; Yowang, A.; Takahashi, A.; Kubo, M.; Shirakata, Y.; Ikezawa, Z.; Iijima, M.; Shiohara, T.; Hashimoto, K.; et al. Genome-wide association study identifies HLA-A*3101 allele as a genetic risk factor for carbamazepine-induced cutaneous adverse drug reactions in Japanese population. Hum. Mol. Genet. 2011, 20, 1034–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browning, S.R.; Browning, B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Wang, J.; Bachtiar, M.; Chong, S.S.; Lee, C.G.L. Architecture of polymorphisms in the human genome reveals functionally important and positively selected variants in immune response and drug transporter genes. Hum. Genom. 2018, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Klein, K.; Saussele, T.; Blievernicht, J.; Hofmann, M.H.; Schwab, M. Polymorphic CYP2B6: Molecular mechanisms and emerging clinical significance. Pharmacogenomics 2007, 8, 743–759. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Batzoglou, S.; Sidow, A.; Zhang, L. HAPDeNovo: A haplotype-based approach for filtering and phasing de novo mutations in linked read sequencing data. BMC Genom. 2018, 19, 467. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M.; Sim, S.C. Pharmacogenetic biomarkers as tools for improved drug therapy; emphasis on the cytochrome P450 system. Biochem. Biophys. Res. Commun. 2010, 396, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ingelman-Sundberg, M.; Lauschke, V.M. Worldwide distribution of cytochrome P450 alleles: A meta-analysis of population-scale sequencing projects. Clin. Pharmacol. Ther. 2017, 102, 688–700. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hadley, D.; Liu, R.; Glessner, J.; Grant, S.F.; Hakonarson, H.; Bucan, M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007, 17, 1665–1674. [Google Scholar] [CrossRef] [Green Version]
- Colella, S.; Yau, C.; Taylor, J.M.; Mirza, G.; Butler, H.; Clouston, P.; Bassett, A.S.; Seller, A.; Holmes, C.C.; Ragoussis, J. QuantiSNP: An Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007, 35, 2013–2025. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Wright, F.A.; Tang, Z.; Nordgard, S.H.; Van Loo, P.; Yu, T.; Kristensen, V.N.; Perou, C.M. Integrated study of copy number states and genotype calls using high-density SNP arrays. Nucleic Acids Res. 2009, 37, 5365–5377. [Google Scholar] [CrossRef]
- Darvishi, K. Application of Nexus copy number software for CNV detection and analysis. Curr. Protoc. Hum. Genet. 2010. [Google Scholar] [CrossRef] [PubMed]
- Seiser, E.L.; Innocenti, F. Hidden Markov Model-Based CNV Detection Algorithms for Illumina Genotyping Microarrays. Cancer Inform. 2014, 13, 77–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellinger, A.E.; Saw, S.M.; Goh, L.K.; Seielstad, M.; Young, T.L.; Li, Y.J. Comparative analyses of seven algorithms for copy number variant identification from single nucleotide polymorphism arrays. Nucleic Acids Res. 2010, 38, e105. [Google Scholar] [CrossRef]
- Fromer, M.; Moran, J.L.; Chambert, K.; Banks, E.; Bergen, S.E.; Ruderfer, D.M.; Handsaker, R.E.; McCarroll, S.A.; O’Donovan, M.C.; Owen, M.J.; et al. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am. J. Hum. Genet. 2012, 91, 597–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krumm, N.; Sudmant, P.H.; Ko, A.; O’Roak, B.J.; Malig, M.; Coe, B.P.; Quinlan, A.R.; Nickerson, D.A.; Eichler, E.E. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012, 22, 1525–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GoldenHelix VarSeq. Available online: https://www.goldenhelix.com/products/VarSeq/ (accessed on 25 November 2020).
- Abyzov, A.; Urban, A.E.; Snyder, M.; Gerstein, M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011, 21, 974–984. [Google Scholar] [CrossRef] [Green Version]
- Yao, R.; Zhang, C.; Yu, T.; Li, N.; Hu, X.; Wang, X.; Wang, J.; Shen, Y. Evaluation of three read-depth based CNV detection tools using whole-exome sequencing data. Mol. Cytogenet. 2017, 10, 30. [Google Scholar] [CrossRef] [Green Version]
- Tremmel, R.; Klein, K.; Battke, F.; Fehr, S.; Winter, S.; Scheurenbrand, T.; Schaeffeler, E.; Biskup, S.; Schwab, M.; Zanger, U.M. Copy number variation profiling in pharmacogenes using panel-based exome resequencing and correlation to human liver expression. Hum. Genet. 2019, 139, 137–149. [Google Scholar] [CrossRef]
- Gaedigk, A. Complexities of CYP2D6 gene analysis and interpretation. Int. Rev. Psychiatry 2013, 25, 534–553. [Google Scholar] [CrossRef]
- Gaedigk, A.; Jaime, L.K.; Bertino, J.S., Jr.; Berard, A.; Pratt, V.M.; Bradfordand, L.D.; Leeder, J.S. Identification of Novel CYP2D7-2D6 Hybrids: Non-Functional and Functional Variants. Front. Pharmacol. 2010, 1, 121. [Google Scholar] [CrossRef] [Green Version]
- Lauschke, V.M.; Milani, L.; Ingelman-Sundberg, M. Pharmacogenomic Biomarkers for Improved Drug Therapy-Recent Progress and Future Developments. AAPS J. 2017, 20, 4. [Google Scholar] [CrossRef] [PubMed]
- Kozyra, M.; Ingelman-Sundberg, M.; Lauschke, V.M. Rare genetic variants in cellular transporters, metabolic enzymes, and nuclear receptors can be important determinants of interindividual differences in drug response. Genet. Med. 2017, 19, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, A.S.; Tabor, H.K.; Johnson, A.D.; Snively, B.M.; Assimes, T.L.; Auer, P.L.; Ioannidis, J.P.; Peters, U.; Robinson, J.G.; Sucheston, L.E.; et al. Quantifying rare, deleterious variation in 12 human cytochrome P450 drug-metabolism genes in a large-scale exome dataset. Hum. Mol. Genet. 2014, 23, 1957–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujikura, K.; Ingelman-Sundberg, M.; Lauschke, V.M. Genetic variation in the human cytochrome P450 supergene family. Pharm. Genom. 2015, 25, 584–594. [Google Scholar] [CrossRef]
- Drogemoller, B.I.; Wright, G.E.; Warnich, L. Considerations for rare variants in drug metabolism genes and the clinical implications. Expert Opin. Drug Metab. Toxicol. 2014, 10, 873–884. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M.; Mkrtchian, S.; Zhou, Y.; Lauschke, V.M. Integrating rare genetic variants into pharmacogenetic drug response predictions. Hum. Genom. 2018, 12, 26. [Google Scholar] [CrossRef]
- Lauschke, V.M.; Ingelman-Sundberg, M. Requirements for comprehensive pharmacogenetic genotyping platforms. Pharmacogenomics 2016, 17, 917–924. [Google Scholar] [CrossRef]
- Lauschke, V.M.; Ingelman-Sundberg, M. How to Consider Rare Genetic Variants in Personalized Drug Therapy. Clin. Pharmacol. Ther. 2018, 103, 745–748. [Google Scholar] [CrossRef]
- Li, B.; Seligman, C.; Thusberg, J.; Miller, J.L.; Auer, J.; Whirl-Carrillo, M.; Capriotti, E.; Klein, T.E.; Mooney, S.D. In silico comparative characterization of pharmacogenomic missense variants. BMC Genom. 2014, 15 (Suppl. S4), S4. [Google Scholar] [CrossRef] [Green Version]
- Han, S.M.; Park, J.; Lee, J.H.; Lee, S.S.; Kim, H.; Han, H.; Kim, Y.; Yi, S.; Cho, J.Y.; Jang, I.J.; et al. Targeted Next-Generation Sequencing for Comprehensive Genetic Profiling of Pharmacogenes. Clin. Pharmacol. Ther. 2017, 101, 396–405. [Google Scholar] [CrossRef]
- Hao, D.; Xiao, P.; Chen, S. Phenotype prediction of nonsynonymous single nucleotide polymorphisms in human phase II drug/xenobiotic metabolizing enzymes: Perspectives on molecular evolution. Sci. China Life Sci. 2010, 53, 1252–1262. [Google Scholar] [CrossRef] [PubMed]
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Klein, T.E.; Korf, B.R.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 2017, 19, 249–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonsalves, S.G.; Dirksen, R.T.; Sangkuhl, K.; Pulk, R.; Alvarellos, M.; Vo, T.; Hikino, K.; Roden, D.; Klein, T.E.; Poler, S.M.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for the Use of Potent Volatile Anesthetic Agents and Succinylcholine in the Context of RYR1 or CACNA1S Genotypes. Clin. Pharmacol. Ther. 2019, 105, 1338–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]


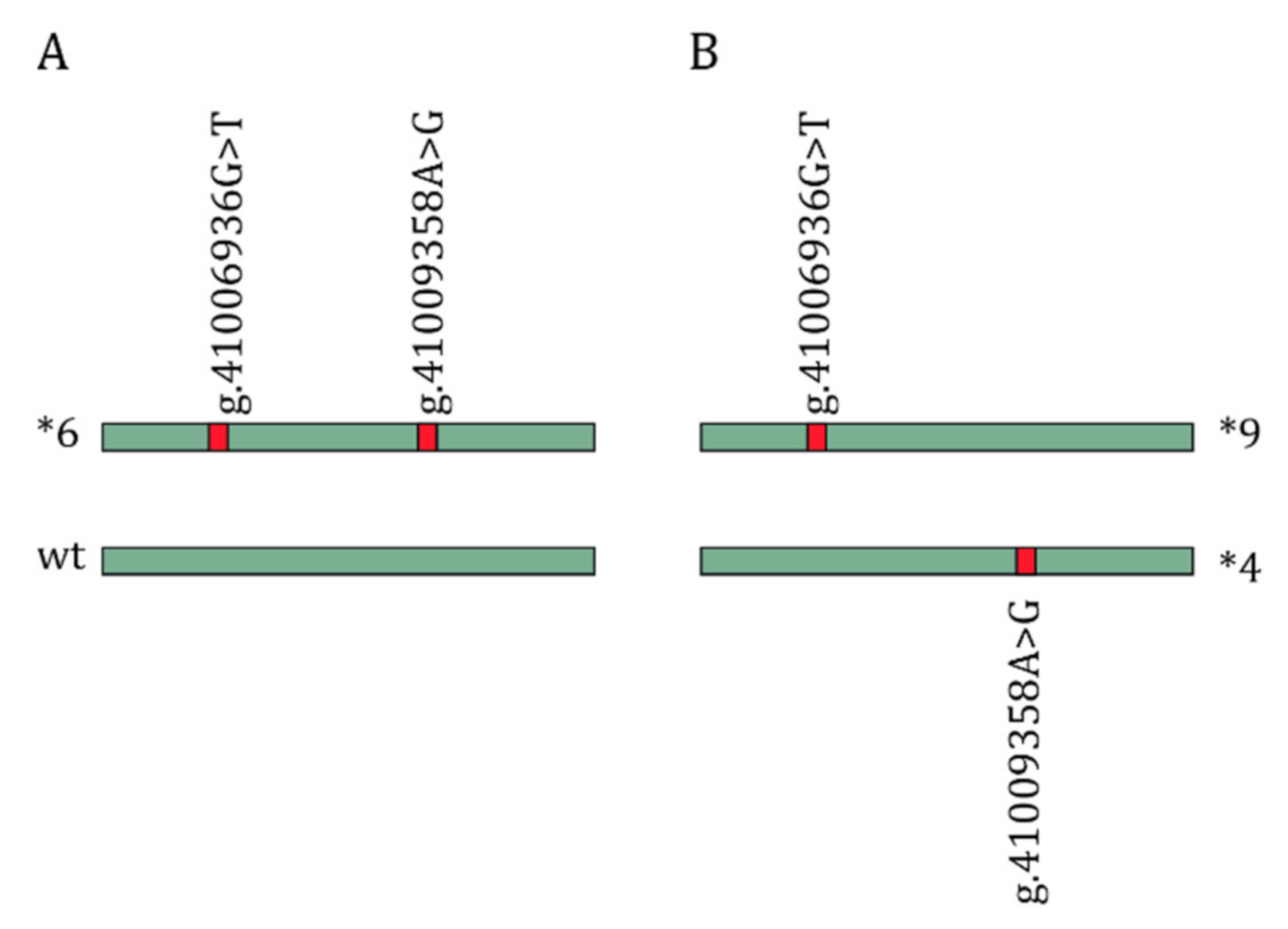{kind=link}
| Protein | Gene | Related Drugs | Locus Size (bp) | Rare Variants, n (% of Known Variants) | Part of Locus Defined as Complex, %(bp) | |
|---|---|---|---|---|---|---|
| CPIC | DPWG | |||||
| CACNA1S | CACNA1S | 7 | - | 73,055 | 2520 (98%) | 33.3 |
| CFTR | CFTR | 1 | - | 250,187 | 1684 (99%) | 42.2 |
| CYP2B6 | CYP2B6 | 1 | 1 | 27,149 | 761 (98%) | 100.0 |
| CYP2C9 | CYP2C9 | 10 | 2 | 50,734 | 632 (98%) | 72.0 |
| CYP2C19 | CYP2C19 | 15 | 10 | 90,525 | 712 (99%) | 83.6 |
| CYP2D6 | CYP2D6 | 14 | 21 | 4408 | 992 (97%) | 100.0 |
| CYP3A5 | CYP3A5 | 1 | 1 | 31,833 | 643 (98%) | 49.4 |
| CYP4F2 | CYP4F2 | 1 | - | 20,098 | 766 (97%) | 51.4 |
| DPD | DPYD | 2 | 4 | 917,258 | 1211 (98%) | 40.0 |
| FACT. V LEIDEN | FACT. V LEIDEN | - | 1 * | 72,423 | 1679 (97%) | 41.9 |
| G6PD | G6PD | 1 | - | 16,183 | 465 (98%) | 36.4 |
| HLA-A | HLA-A | 2 | 1 | 4625 | 423 (71%) | 100.0 |
| HLA-B | HLA-B | 6 | 7 | 87,698 | 308 (78%) | 62.1 |
| IFNL3 | IFNL3 | 2 | - | 1577 | 317 (95%) | 100.0 |
| IFNL4 | IFNL4 | 2 | - | 3543 | 404 (97%) | 100.0 |
| NUDT15 | NUDT15 | 3 | 3 | 9656 | 244 (99%) | 64.7 |
| RYR-1 | RYR1 | 7 | - | 153,866 | 6584 (98%) | 51.4 |
| SLCO1B1 | SLCO1B1 | 1 | 2 | 108,045 | 951 (96%) | 69.6 |
| TPMT | TPMT | 3 | 3 | 26,764 | 346 (97%) | 52.3 |
| UGT1A1 | UGT1A1 | 1 | 1 | 13,052 | 470 (99%) | 40.3 |
| VKORC1 | VKORC1 | 1 | 3 | 5139 | 370 (98%) | 41.8 |
| SNV Panel | Short-Read Seq | Long-Read Seq | ||||||
|---|---|---|---|---|---|---|---|---|
| PGx Panel | Whole Genome Panel | PGx Panel | WES | WGS | PGx Panel | WGS | ||
| Turnaround Time Wetlab * | ++ | + | + | + | +/- | - | -- | |
| Haplotype phasing | Computational | - | +/- | + | +/- | + | ++ | ++ |
| Direct | - | - | - | - | - | ++ | ++ | |
| Imputation | - | +/- | +/- | +/- | NA | NA | NA | |
| Coverage of PGx variation | + | +/- | ++ | +/- | ++ | ++ | ++ | |
| Detection of rare variants † | + | + | ++ | +/- | ++ | ++ | ++ | |
| Detection of variants outside the predefined gene/variant panel | -- | -- | -/+ | -/+ | ++ | ++ | ++ | |
| Detection of structural and complex variants | -- | -- | + | +/- | + | ++ | ++ | |
| Turnaround time data processing * | ++ | ++ | + | + | +/- | - | -- | |
| Costs ‡ [29] | Investment | ++ | + | - | - | - | - | - |
| Running costs per sample | +/- | ++ | +/- | +/- | - | +/- | - | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Lee, M.; Kriek, M.; Guchelaar, H.-J.; Swen, J.J. Technologies for Pharmacogenomics: A Review. Genes 2020, 11, 1456. https://doi.org/10.3390/genes11121456
van der Lee M, Kriek M, Guchelaar H-J, Swen JJ. Technologies for Pharmacogenomics: A Review. Genes. 2020; 11(12):1456. https://doi.org/10.3390/genes11121456
Chicago/Turabian Stylevan der Lee, Maaike, Marjolein Kriek, Henk-Jan Guchelaar, and Jesse J. Swen. 2020. "Technologies for Pharmacogenomics: A Review" Genes 11, no. 12: 1456. https://doi.org/10.3390/genes11121456
APA Stylevan der Lee, M., Kriek, M., Guchelaar, H. -J., & Swen, J. J. (2020). Technologies for Pharmacogenomics: A Review. Genes, 11(12), 1456. https://doi.org/10.3390/genes11121456