Repetitive DNA Restructuring Across Multiple Nicotiana Allopolyploidisation Events Shows a Lack of Strong Cytoplasmic Bias in Influencing Repeat Turnover

 , and
, and
Abstract
:1. Introduction
- How do repeat dynamics vary across polyploidisation events of different ages?
- Is there a higher contribution of maternal subgenome DNA to the allopolyploid genome, reflected in repetitive element abundances?
- If present, do these biases vary with the age of the allopolyploid, and/or phylogenetic distance of progenitor subgenomes?
2. Materials and Methods
2.1. Illumina Sequencing Data
2.2. Clustering of Read Data
2.3. Statistical Analyses
3. Results
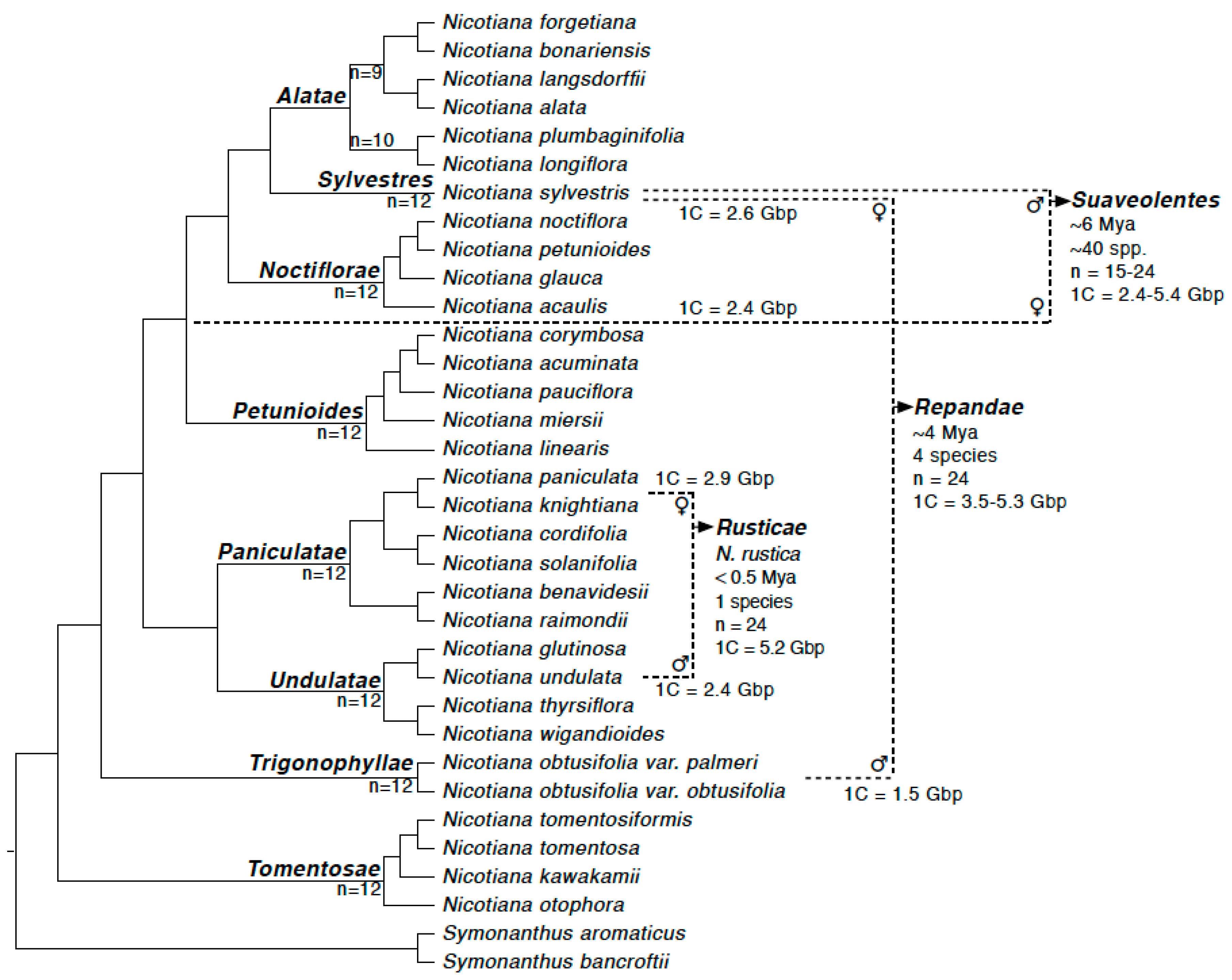
3.1. Repeat Dynamics in Polyploids of 0.5–6 Ma
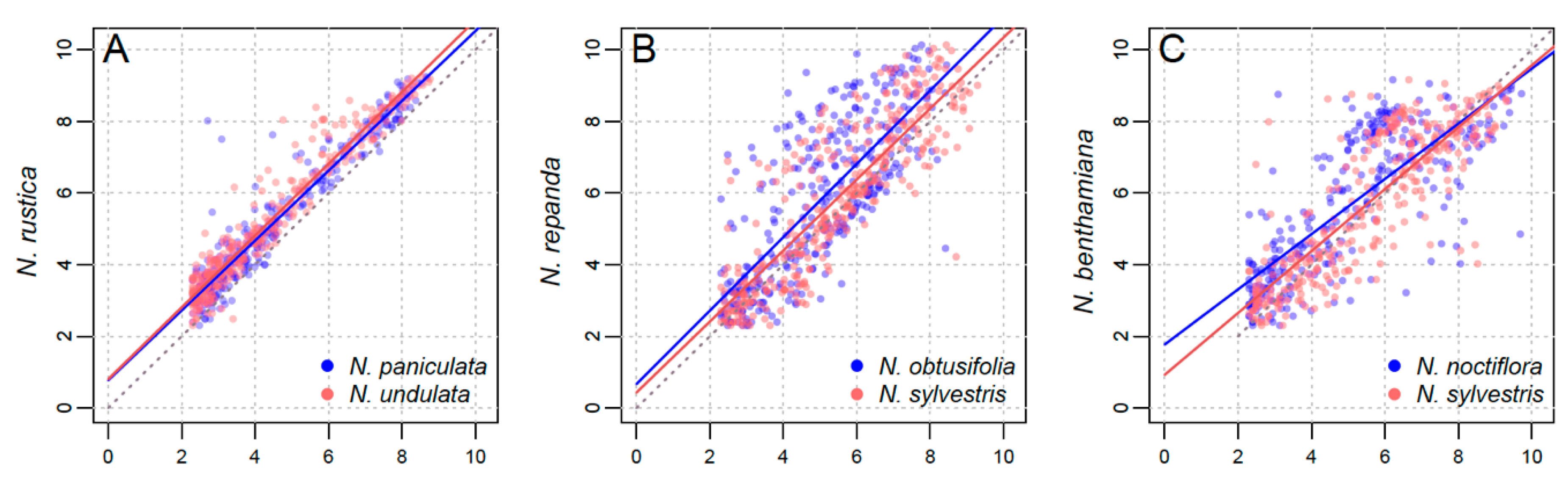
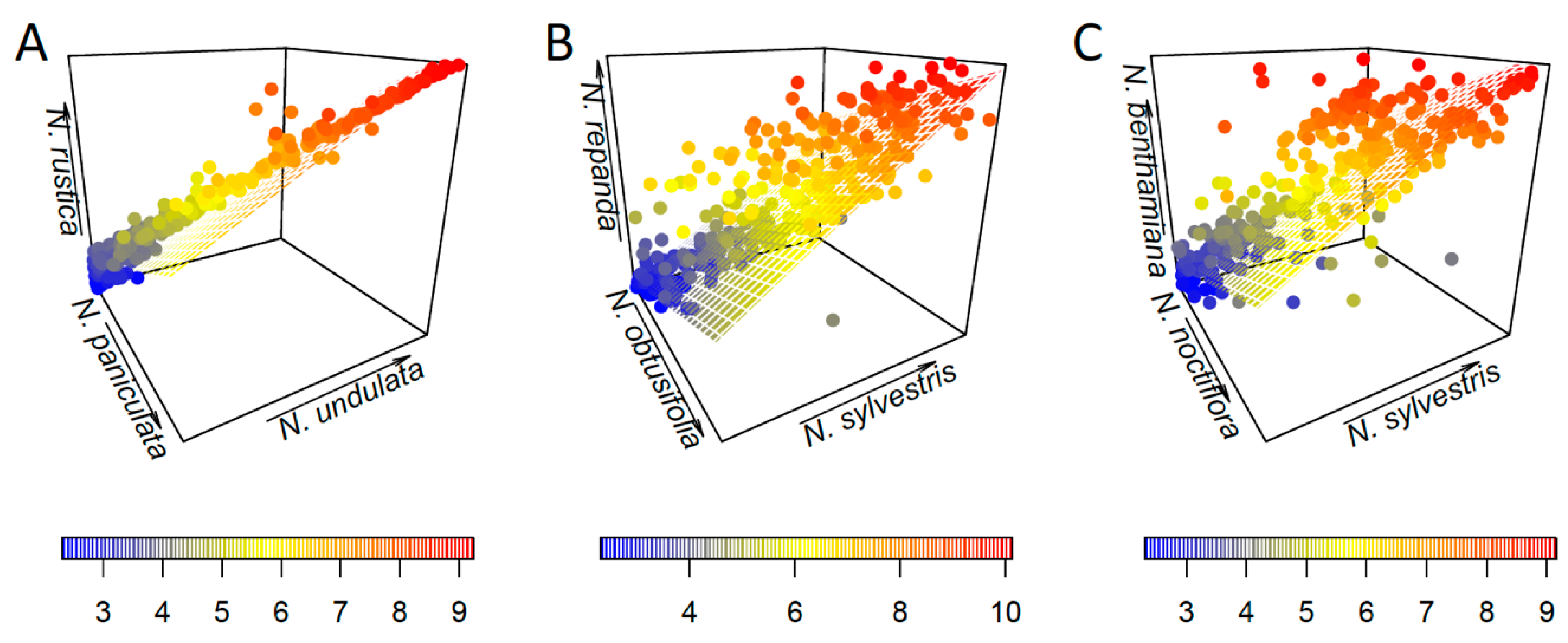
3.2. Parental Contribution to Allotetraploid Genomes
4. Discussion
4.1. Repeat Restructuring across 0.5–6 Ma Timescales
4.2. Lack of a Clear Parental Bias in Repeat Loss/Retention
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wendel, J.F. The wondrous cycles of polyploidy in plants. Am. J. Bot. 2015, 102, 1753–1756. [Google Scholar] [CrossRef] [Green Version]
- Wood, T.E.; Takebayashi, N.; Barker, M.S.; Mayrose, I.; Greenspoon, P.B.; Rieseberg, L.H. The frequency of polyploid speciation in vascular plants. Proc. Natl. Acad. Sci. USA 2009, 106, 13875–13879. [Google Scholar] [CrossRef] [Green Version]
- Mayrose, I.; Zhan, S.H.; Rothfels, C.J.; Magnuson-Ford, K.; Barker, M.S.; Rieseberg, L.H.; Otto, S.P. Recently formed polyploid plants diversify at lower rates. Science 2011, 333, 1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landis, J.B.; Soltis, D.E.; Li, Z.; Marx, H.E.; Barker, M.S.; Tank, D.C.; Soltis, P.S. Impact of whole-genome duplication events on diversification rates in angiosperms. Am. J. Bot. 2018, 105, 348–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eric Schranz, M.; Mohammadin, S.; Edger, P.P. Ancient whole genome duplications, novelty and diversification: The WGD Radiation Lag-Time Model. Curr. Opin. Plant Biol. 2012, 15, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Tank, D.C.; Eastman, J.M.; Pennell, M.W.; Soltis, P.S.; Soltis, D.E.; Hinchliff, C.E.; Brown, J.W.; Sessa, E.B.; Harmon, L.J. Nested radiations and the pulse of angiosperm diversification: Increased diversification rates often follow whole genome duplications. New Phytol. 2015, 207, 454–467. [Google Scholar] [CrossRef] [Green Version]
- Dodsworth, S.; Chase, M.W.; Leitch, A.R. Is post-polyploidization diploidization the key to the evolutionary success of angiosperms? Bot. J. Linn. Soc. 2016, 180, 1–5. [Google Scholar] [CrossRef]
- Lim, K.Y.; Matyasek, R.; Kovarik, A.; Leitch, A.R. Genome evolution in allotetraploid Nicotiana. Biol. J. Linn. Soc. 2004, 82, 599–606. [Google Scholar] [CrossRef]
- Renny-Byfield, S.; Chester, M.; Kovarik, A.; Le Comber, S.C.; Grandbastien, M.-A.; Deloger, M.; Nichols, R.A.; Macas, J.; Novak, P.; Chase, M.W.; et al. Next generation sequencing reveals genome downsizing in allotetraploid Nicotiana tabacum, predominantly through the elimination of paternally derived repetitive DNAs. Mol. Biol. Evol. 2011, 28, 2843–2854. [Google Scholar] [CrossRef] [Green Version]
- Renny-Byfield, S.; Kovařík, A.; Chester, M.; Nichols, R.A.; Macas, J.; Novák, P.; Leitch, A.R. Independent, rapid and targeted loss of highly repetitive DNA in natural and synthetic allopolyploids of Nicotiana tabacum. PLoS ONE 2012, 7, e36963. [Google Scholar] [CrossRef] [Green Version]
- Dodsworth, S.; Jang, T.S.; Struebig, M.; Chase, M.W.; Weiss-Schneeweiss, H.; Leitch, A.R. Genome-wide repeat dynamics reflect phylogenetic distance in closely related allotetraploid Nicotiana (Solanaceae). Plant Syst. Evol. 2017, 303, 1013–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarkson, J.J.; Lim, K.Y.; Kovarik, A.; Chase, M.W.; Knapp, S.; Leitch, A.R. Long-term genome diploidization in allopolyploid Nicotiana section Repandae (Solanaceae). New Phytol. 2005, 168, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Renny-Byfield, S.; Kovarik, A.; Kelly, L.J.; Macas, J.; Novak, P.; Chase, M.W.; Nichols, R.A.; Pancholi, M.R.; Grandbastien, M.-A.; Leitch, A.R. Diploidization and genome size change in allopolyploids is associated with differential dynamics of low- and high-copy sequences. Plant J. 2013, 74, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, J.J.; Knapp, S.; Garcia, V.F.; Olmstead, R.G.; Leitch, A.R.; Chase, M.W. Phylogenetic relationships in Nicotiana (Solanaceae) inferred from multiple plastid DNA regions. Mol. Phylogenet. Evol. 2004, 33, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, J.J.; Kelly, L.J.; Leitch, A.R.; Knapp, S.; Chase, M.W. Nuclear glutamine synthetase evolution in Nicotiana: Phylogenetics and the origins of allotetraploid and homoploid (diploid) hybrids. Mol. Phylogenet. Evol. 2010, 55, 99–112. [Google Scholar] [CrossRef]
- Kelly, L.J.; Leitch, A.R.; Clarkson, J.J.; Hunter, R.B.; Knapp, S.; Chase, M.W. Intragenic recombination events and evidence for hybrid speciation in Nicotiana (Solanaceae). Mol. Biol. Evol. 2010, 27, 781–799. [Google Scholar] [CrossRef] [Green Version]
- Kelly, L.J.; Leitch, A.R.; Clarkson, J.J.; Knapp, S.; Chase, M.W. Reconstructing the complex evolutionary origin of wild allopolyploid tobaccos (Nicotiana section Suaveolentes). Evolution 2013, 67, 80–94. [Google Scholar] [CrossRef]
- Clarkson, J.J.; Dodsworth, S.; Chase, M.W. Time-calibrated phylogenetic trees establish a lag between polyploidisation and diversification in Nicotiana (Solanaceae). Plant Syst. Evol. 2017, 303, 1001–1012. [Google Scholar] [CrossRef]
- Knapp, S.; Chase, M.W.; Clarkson, J.J. Nomenclatural changes and a new sectional classification in Nicotiana (Solanaceae). Taxon 2004, 53, 73–82. [Google Scholar] [CrossRef]
- Schiavinato, M.; Marcet-houben, M.; Dohm, J.C.; Programme, G.; Barcelona, T. Parental origin of the allotetraploid tobacco Nicotiana benthamiana. Plant J. 2019. [Google Scholar] [CrossRef] [Green Version]
- Dodsworth, S.; Kovarik, A.; Grandbastien, M.-A.; Leitch, I.J.; Leitch, A.R. Repetitive DNA dynamics and polyploidization in the genus Nicotiana (Solanaceae). In The Tobacco Genome; Ivanov, N.V., Ed.; Springer: Basel, Switzerland, 2020. [Google Scholar]
- Leitch, I.J.; Hanson, L.; Lim, K.Y.; Kovarik, A.; Chase, M.W.; Clarkson, J.J.; Leitch, A.R. The ups and downs of genome size evolution in polyploid species of Nicotiana (Solanaceae). Ann. Bot. 2008, 101, 805–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitch, A.R.; Lim, K.Y.; Skalicka, K.; Kovarik, A. Nuclear cytoplasmic interaction hypothesis and the role of translocations in Nicotiana allopolyploids. In Radiation Risk Estimates in Normal and Emergency Situations; Cigma, A.A., Durante, M., Eds.; Springer: Basel, Switzerland, 2006; pp. 319–326. [Google Scholar]
- Sierro, N.; Battey, J.N.D.; Bovet, L.; Liedschulte, V.; Ouadi, S.; Thomas, J.; Broye, H.; Laparra, H.; Vuarnoz, A.; Lang, G.; et al. The impact of genome evolution on the allotetraploid Nicotiana rustica—An intriguing story of enhanced alkaloid production. BMC Genom. 2018, 19, 855. [Google Scholar] [CrossRef] [PubMed]
- Dodsworth, S.; Guignard, M.S.; Christenhusz, M.J.M.; Cowan, R.S.; Knapp, S.; Maurin, O.; Struebig, M.; Leitch, A.R.; Chase, M.W.; Forest, F. Potential of herbariomics for studying repetitive DNA in angiosperms. Front. Ecol. Evol. 2018, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Novák, P.; Neumann, P.; Macas, J. Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinform. 2010, 11, 378. [Google Scholar] [CrossRef] [Green Version]
- Novák, P.; Neumann, P.; Pech, J.; Steinhaisl, J.; Macas, J. RepeatExplorer: A Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 2013, 29, 792–793. [Google Scholar] [CrossRef] [Green Version]
- Development Core Team. A Language and Environment for Statistical Computing; Found. Stat. Comput: Vienna, Austria, 2016; ISBN 3-900051-07-0. [Google Scholar]
- Soetaert, K. Plotting Multi-Dimensional Data; R package: Yerseke, The Netherlands, 2014. [Google Scholar]
- Fox, J.; Weisberg, S.; Adler, D.; Bates, D.M.; Baud-Bovy, G.; Ellison, S.; Firth, D.; Friendly, M.; Gorjanc, G.; Graves, S.; et al. An R Companion to Applied Regression, 2nd ed.; SAGE Publishing: Thousand Oaks, CA, USA, 2014. [Google Scholar]
- Lim, K.Y.; Matyasek, R.; Kovarik, A.; Fulnecek, J.; Leitch, A.R. Molecular cytogenetics and tandem repeat sequence evolution in the allopolyploid Nicotiana rustica compared with diploid progenitors N. paniculata and N. undulata. Cytogenet. Genome Res. 2005, 109, 298–309. [Google Scholar] [CrossRef]
- Matyasek, R.; Lim, K.Y.; Kovarik, A.; Leitch, A.R. Ribosomal DNA evolution and gene conversion in Nicotiana rustica. Heredity 2003, 91, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Chase, M.W.; Christenhusz, M.J.M.; Conran, J.G.; Dodsworth, S.; Medeiros de Assis, F.N.; Felix, L.P.; Fay, M.F. Unexpected diversity of Australian tobacco species (Nicotiana section Suaveolentes, Solanaceae). Curtis Bot. Mag. 2018, 35, 212–227. [Google Scholar] [CrossRef]
- Dodsworth, S. Genome Skimming for Phylogenomics. Ph.D. Thesis, Queen Mary University of London, London, UK, 2015. [Google Scholar]
- Mandáková, T.; Lysak, M.A. Post-polyploid diploidization and diversification through dysploid changes. Curr. Opin. Plant Biol. 2018, 42, 55–65. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polyploid | Parents | Sum of Sq. | F Stat | p-Value |
|---|---|---|---|---|
| N. benthamiana | N. noctiflora, N. sylvestris | 8.617 | 8.138 | 0.0046 |
| N. repanda | N. obtusifolia, N. sylvestris | 14.297 | 13.840 | 0.0002 |
| N. rustica | N. paniculata, N. undulata | 0.491 | 5.451 | 0.0201 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dodsworth, S.; Guignard, M.S.; Pérez-Escobar, O.A.; Struebig, M.; Chase, M.W.; Leitch, A.R. Repetitive DNA Restructuring Across Multiple Nicotiana Allopolyploidisation Events Shows a Lack of Strong Cytoplasmic Bias in Influencing Repeat Turnover. Genes 2020, 11, 216. https://doi.org/10.3390/genes11020216
Dodsworth S, Guignard MS, Pérez-Escobar OA, Struebig M, Chase MW, Leitch AR. Repetitive DNA Restructuring Across Multiple Nicotiana Allopolyploidisation Events Shows a Lack of Strong Cytoplasmic Bias in Influencing Repeat Turnover. Genes. 2020; 11(2):216. https://doi.org/10.3390/genes11020216
Chicago/Turabian StyleDodsworth, Steven, Maïté S. Guignard, Oscar A. Pérez-Escobar, Monika Struebig, Mark W. Chase, and Andrew R. Leitch. 2020. "Repetitive DNA Restructuring Across Multiple Nicotiana Allopolyploidisation Events Shows a Lack of Strong Cytoplasmic Bias in Influencing Repeat Turnover" Genes 11, no. 2: 216. https://doi.org/10.3390/genes11020216
APA StyleDodsworth, S., Guignard, M. S., Pérez-Escobar, O. A., Struebig, M., Chase, M. W., & Leitch, A. R. (2020). Repetitive DNA Restructuring Across Multiple Nicotiana Allopolyploidisation Events Shows a Lack of Strong Cytoplasmic Bias in Influencing Repeat Turnover. Genes, 11(2), 216. https://doi.org/10.3390/genes11020216