Clinical Observations and Treatment Approaches for Scoliosis in Prader–Willi Syndrome



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Clinical Observations and Treatment
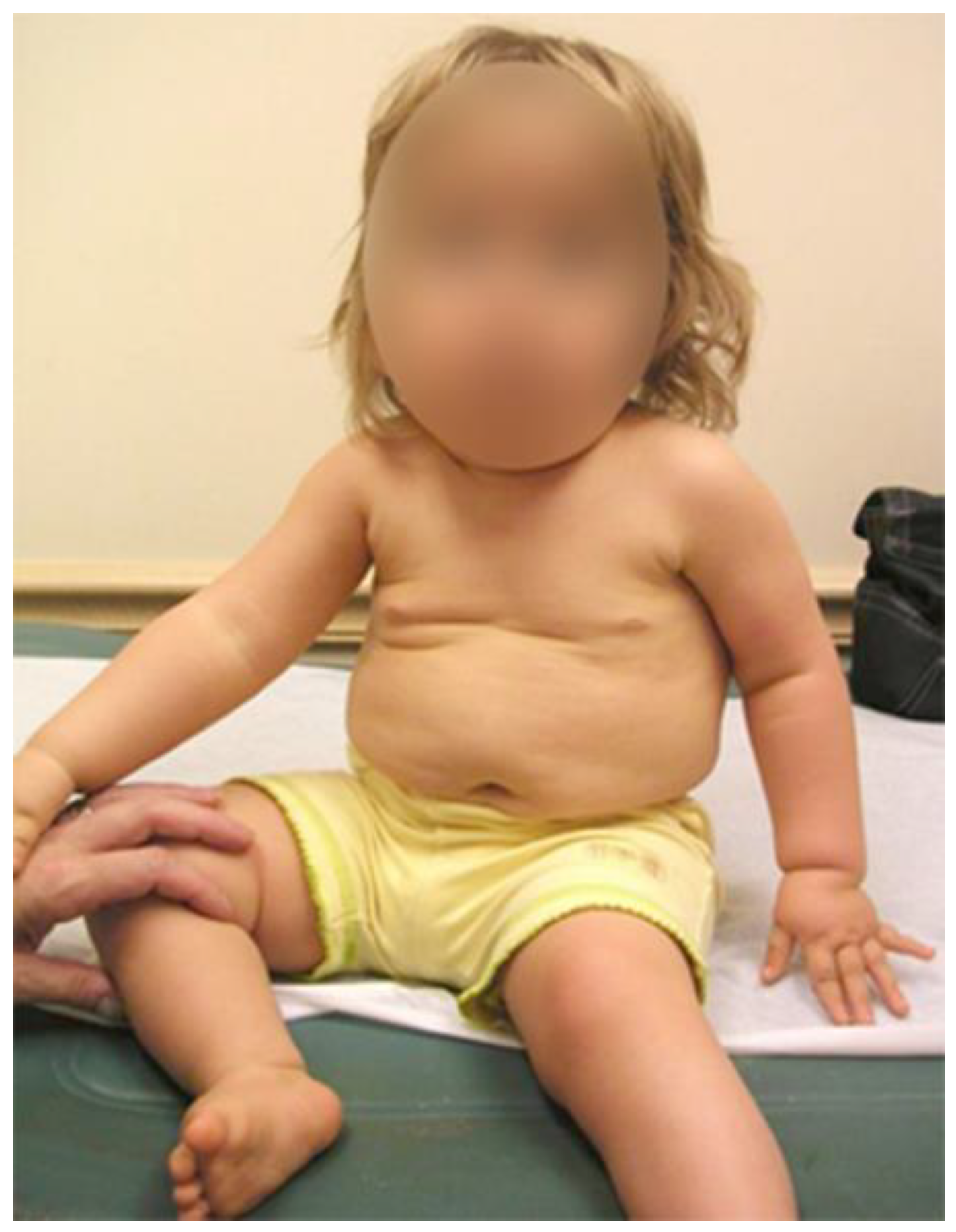
2.1. Clinical Experiences and Treatment for Scoliosis in Prader–Willi Syndrome
2.1.1. Physical Therapy
2.1.2. Screening
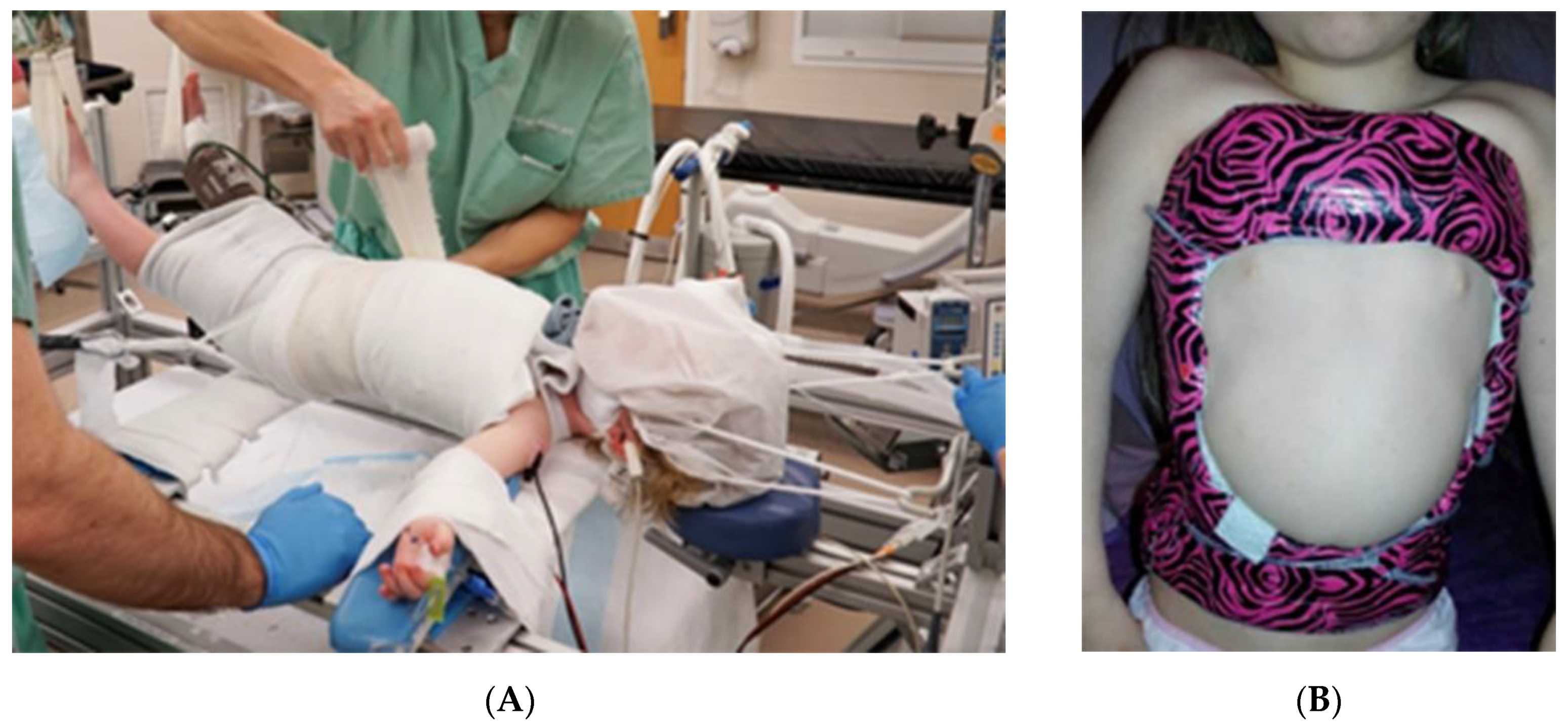
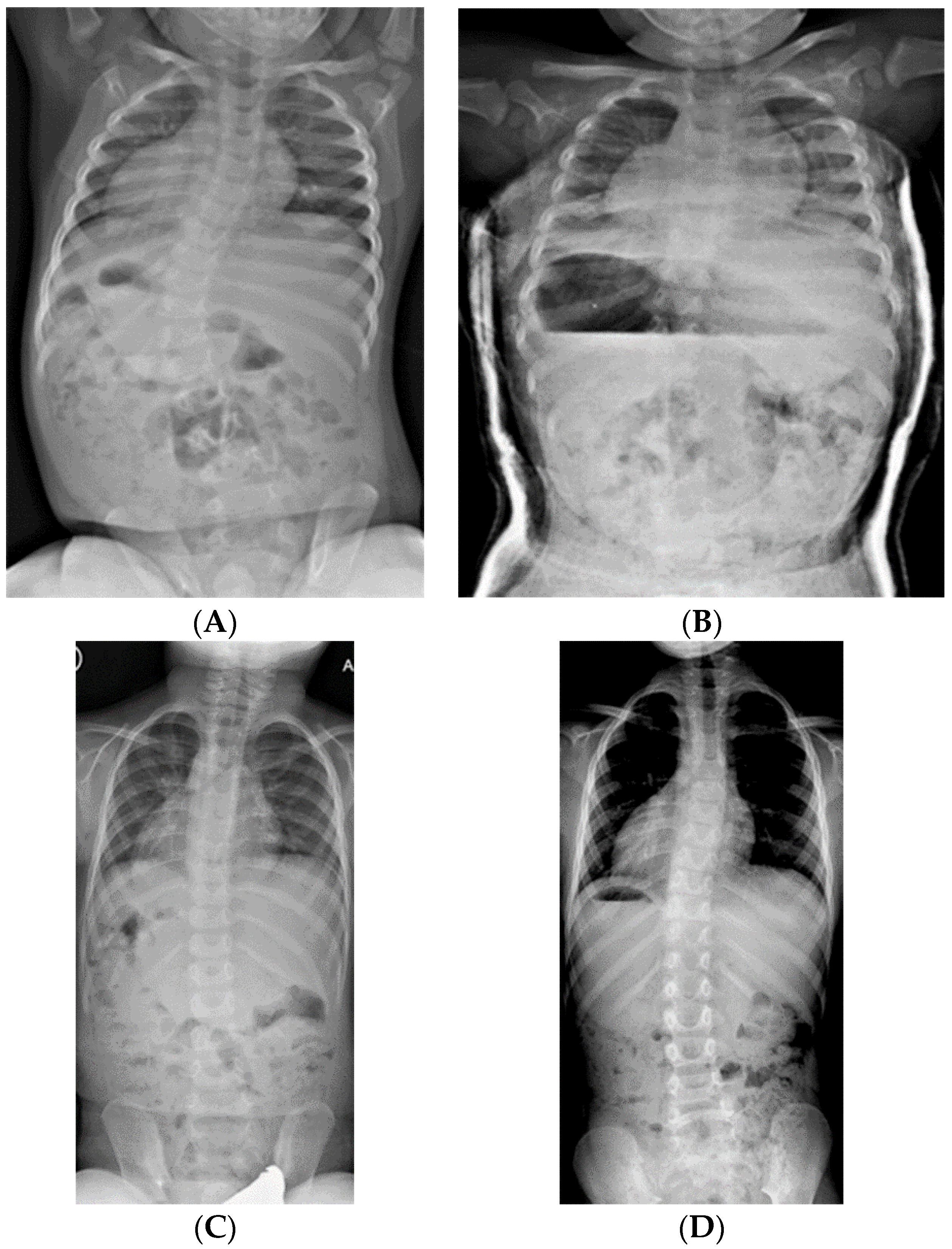
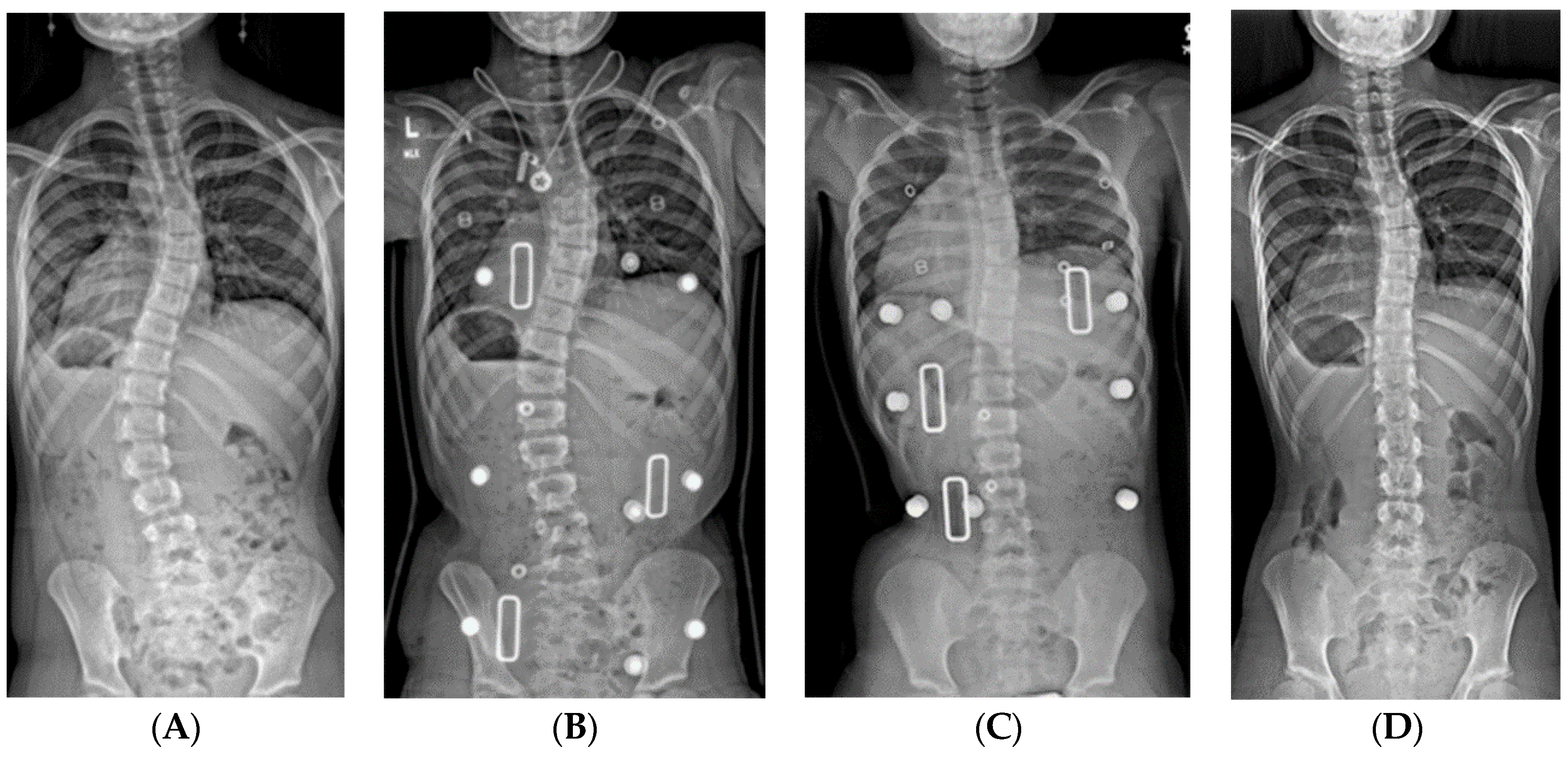
2.1.3. Serial Spinal Casting
2.1.4. Bracing
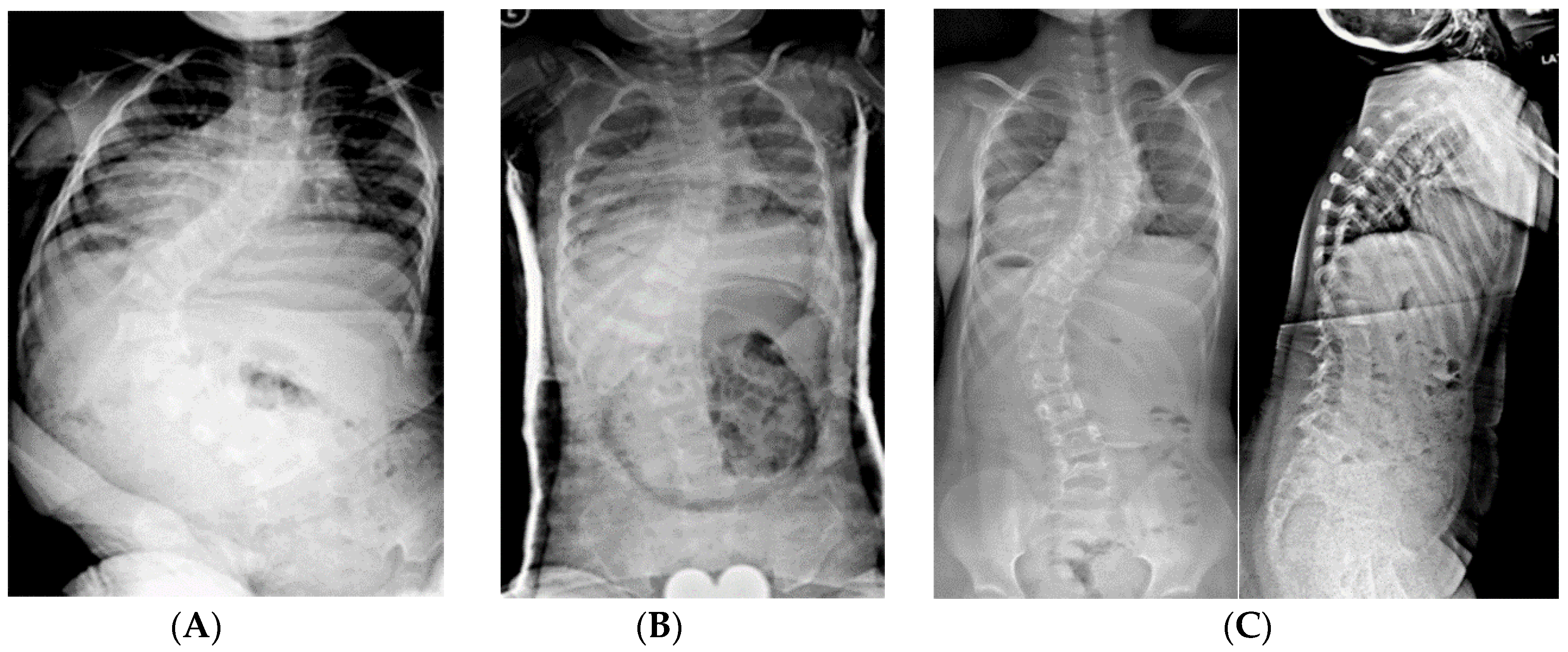
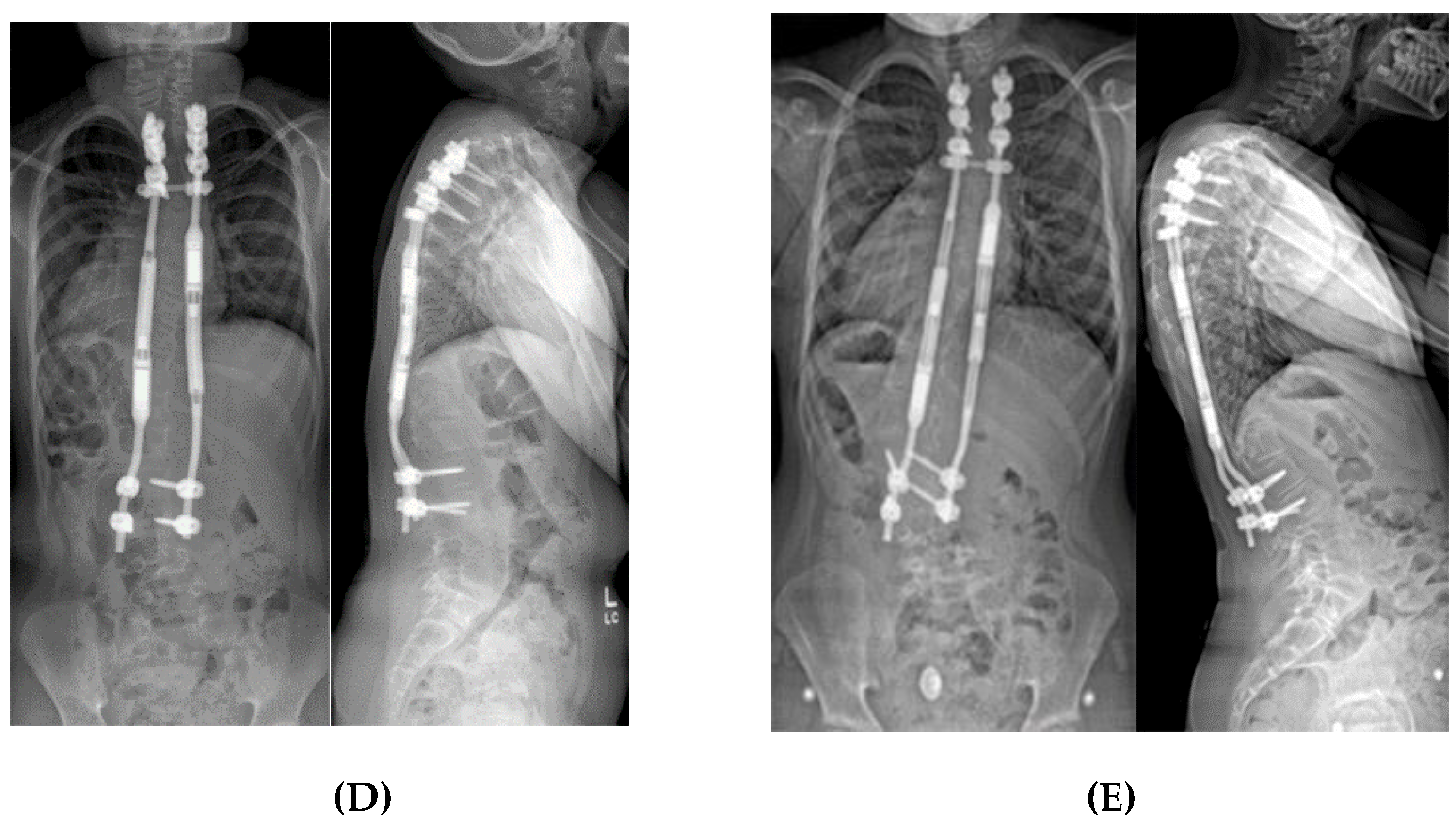
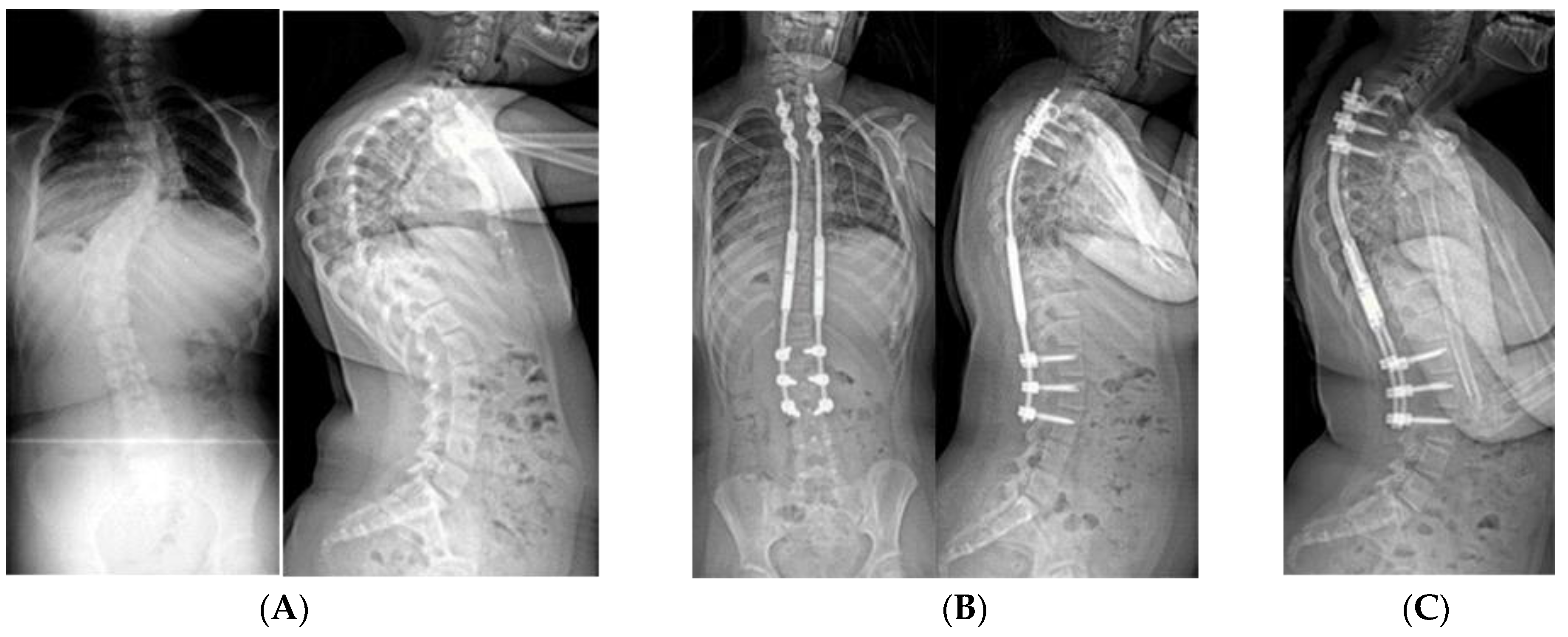
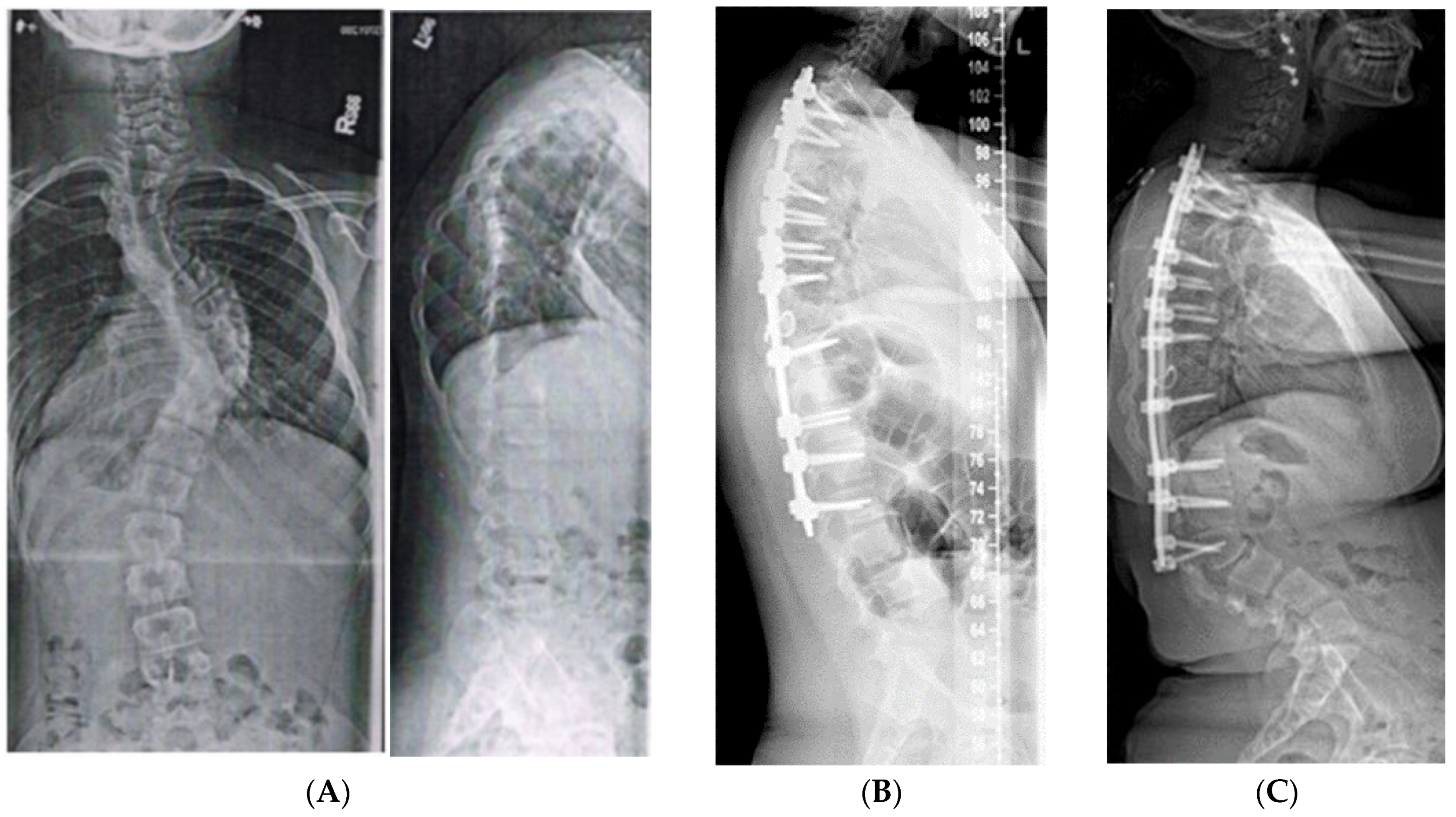
2.1.5. Spine Surgery
2.1.5.1. Complications
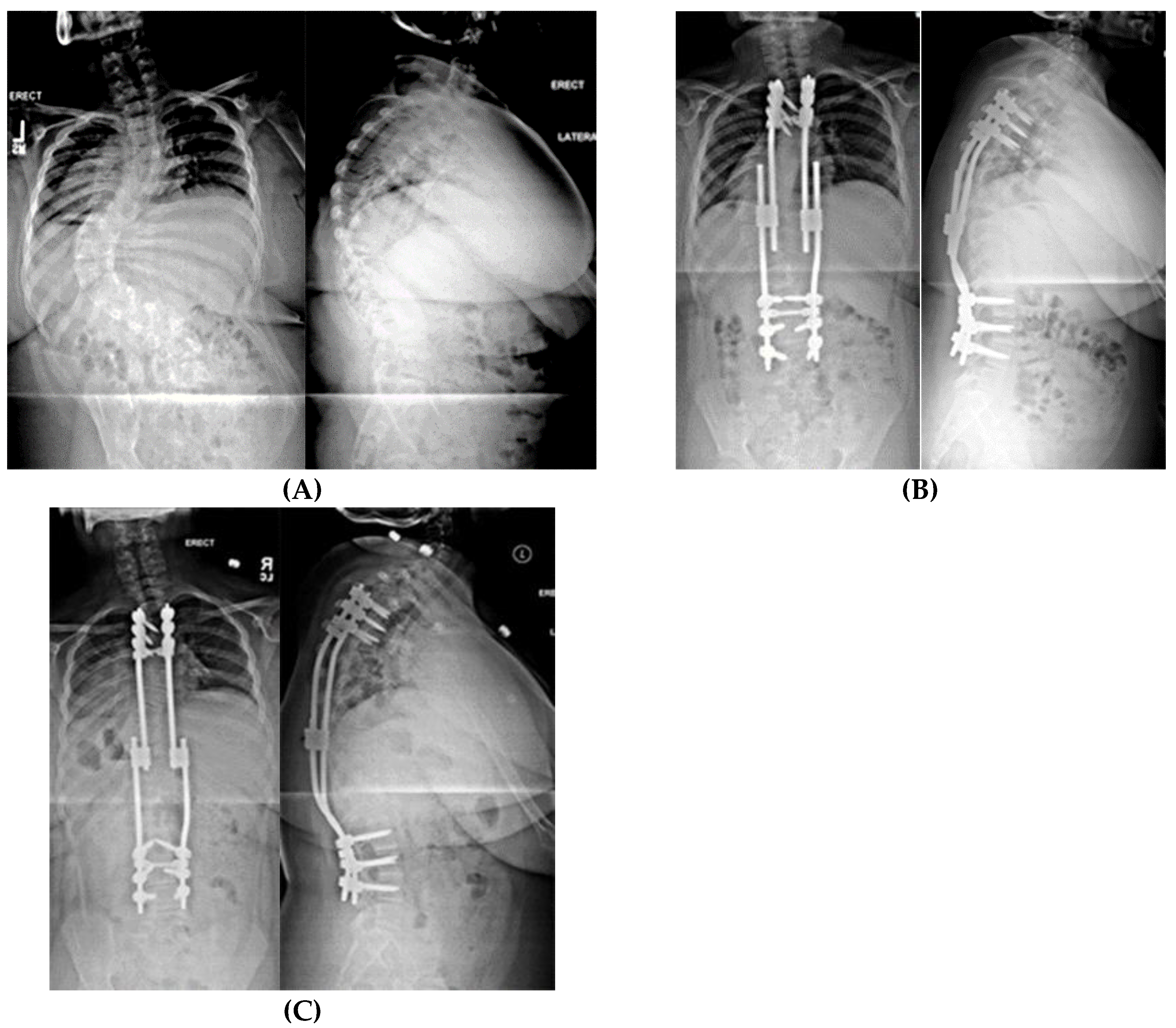
2.1.5.2. Non-Fusion Spinal Instrumentation with Expandable Implants
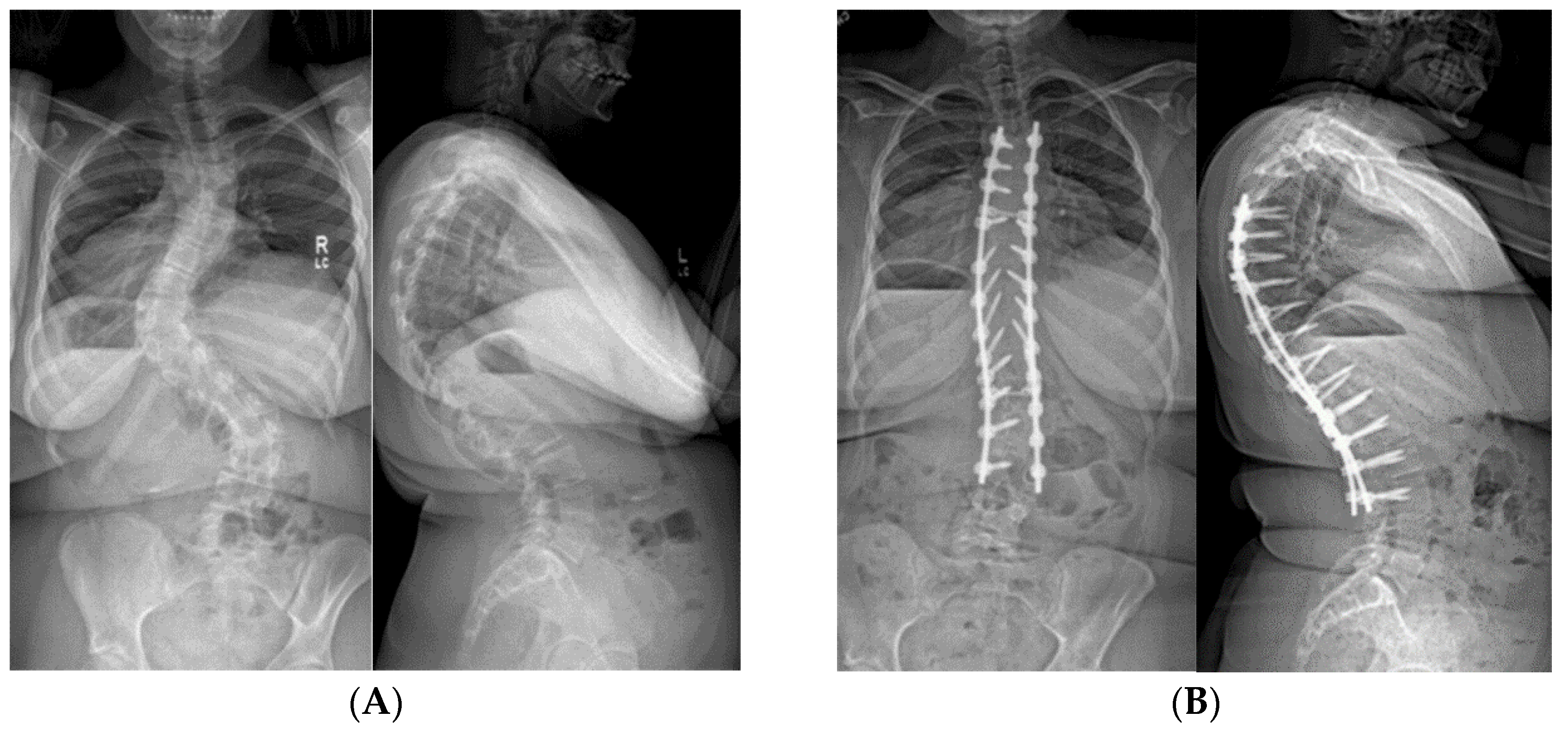
2.1.5.3. Spinal Fusion
3. Discussion
Funding
Acknowledgments
Conflicts of Interest
References
- Butler, M.G.; Hanchett, J.M.; Thompson, T. Clinical findings and natural history of Prader-Willi syndrome. In Management of Prader-Willi Syndrome, 3rd ed.; Butler, M.G., Lee, P.D.K., Whitman, Y.W., Eds.; Springer: New York, NY, USA, 2006; pp. 3–48. [Google Scholar]
- Butler, M.G.; Thompson, T. Prader-Willi syndrome: Clinical and genetic findings. Endocrinologist 2000, 10, 3S–16S. [Google Scholar] [CrossRef]
- Butler, M.G. Prader-Willi Syndrome: Current understanding of cause and diagnosis. Am. J. Med. Genet. 1990, 35, 319–332. [Google Scholar] [CrossRef] [Green Version]
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.G.; Hartin, S.N.; Hossain, W.A.; Manzardo, A.M.; Kimonis, V.; Dykens, E.; Gold, J.A.; Kim, S.J.; Weisensel, N.; Tamura, R.; et al. Molecular genetic classification in Prader-Willi syndrome: A multisite cohort study. J. Med. Genet. 2019, 56, 149–153. [Google Scholar] [CrossRef]
- Butler, M.G.; Bittel, D.C.; Kibiryeva, N.; Talebizadeh, Z.; Thompson, T. Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics 2004, 113, 565–573. [Google Scholar] [CrossRef]
- Butler, M.G.; Matthews, N.A.; Patel, N.; Surampalli, A.; Gold, J.A.; Khare, M.; Thompson, T.; Cassidy, S.B.; Kimonis, V.E. Impact of genetic subtypes of Prader-Willi syndrome with growth hormone therapy on intelligence and body mass index. Am. J. Med. Genet. A 2019, 179, 1826–1835. [Google Scholar] [CrossRef]
- de Lind van Wijngaarden, R.F.; de Klerk, L.W.; Festen, D.A.; Hokken-Koelega, A.C. Scoliosis in Prader-Willi syndrome: Prevalence, effects of age, gender, body mass index, lean body mass and genotype. Arch. Dis. Child. 2008, 93, 1012–1016. [Google Scholar] [CrossRef] [Green Version]
- Greggi, T.; Martikos, K.; Lolli, F.; Bakaloudis, G.; Di Silvestre, M.; Cioni, A.; Brodano, G.B.; Giacomini, S. Treatment of scoliosis in patients affected with Prader-Willi syndrome using various techniques. Scoliosis 2010, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Holm, V.A.; Laurnen, E.L. Prader-Willi syndrome and scoliosis. Dev. Med. Child. Neurol. 1981, 23, 192–201. [Google Scholar] [CrossRef]
- Kroonen, L.T.; Herman, M.; Pizzutillo, P.D.; Macewen, G.D. Prader-Willi syndrome: Clinical concerns for the orthopaedic surgeon. J. Pediatr. Orthop. 2006, 26, 673–679. [Google Scholar] [CrossRef]
- Nagai, T.; Obata, K.; Ogata, T.; Murakami, N.; Katada, Y.; Yoshino, A.; Sakazume, S.; Tomita, Y.; Sakuta, R.; Niikawa, N. Growth hormone therapy and scoliosis in patients with Prader-Willi syndrome. Am. J. Med. Genet. A 2006, 140, 1623–1627. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Murakami, N.; Iida, T.; Ozeki, S.; Asano, S.; Nohara, Y.; Nagai, T. The characteristics of scoliosis in Prader-Willi syndrome (PWS): Analysis of 58 scoliosis patients with PWS. J. Orthop. Sci. 2015, 20, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Odent, T.; Accadbled, F.; Koureas, G.; Cournot, M.; Moine, A.; Diene, G.; Molinas, C.; Pinto, G.; Tauber, M.; Gomes, B.; et al. Scoliosis in patients with Prader-Willi syndrome. Pediatrics 2008, 122, e499–e503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghergan, A.; Coupaye, M.; Leu-Semenescu, S.; Attali, V.; Oppert, J.M.; Arnulf, I.; Poitou, C.; Redolfi, S. Prevalence and phenotype of sleep disorders in 60 adults with Prader-Willi syndrome. Sleep 2017, 40. [Google Scholar] [CrossRef]
- Sedky, K.; Bennett, D.S.; Pumariega, A. Prader-Willi syndrome and obstructive sleep apnea: Co-occurrence in the pediatric population. J. Clin. Sleep Med. 2014, 10, 403–409. [Google Scholar] [CrossRef]
- Tan, H.L.; Urquhart, D.S. Respiratory complications in children with Prader Willi syndrome. Paediatr. Respir. Rev. 2017, 22, 52–59. [Google Scholar] [CrossRef]
- Gurd, A.R.; Thompson, T.R. Scoliosis in Prader-Willi syndrome. J. Pediatr. Orthop. 1981, 1, 317–320. [Google Scholar] [CrossRef]
- Hakonarson, H.; Moskovitz, J.; Daigle, K.L.; Cassidy, S.B.; Cloutier, M.M. Pulmonary function abnormalities in Prader-Willi syndrome. J. Pediatr. 1995, 126, 565–570. [Google Scholar] [CrossRef]
- Pacoricona Alfaro, D.L.; Lemoine, P.; Ehlinger, V.; Molinas, C.; Diene, G.; Valette, M.; Pinto, G.; Coupaye, M.; Poitou-Bernert, C.; Thuilleaux, D.; et al. Causes of death in Prader-Willi syndrome: Lessons from 11 years’ experience of a national reference center. Orphanet J. Rare Dis. 2019, 14, 238. [Google Scholar] [CrossRef] [Green Version]
- Proffitt, J.; Osann, K.; McManus, B.; Kimonis, V.E.; Heinemann, J.; Butler, M.G.; Stevenson, D.A.; Gold, J.A. Contributing factors of mortality in Prader-Willi syndrome. Am. J. Med. Genet. A 2019, 179, 196–205. [Google Scholar] [CrossRef]
- Rees, D.; Jones, M.W.; Owen, R.; Dorgan, J.C. Scoliosis surgery in the Prader-Willi syndrome. J. Bone Jt. Surg. Br. 1989, 71, 685–688. [Google Scholar] [CrossRef] [Green Version]
- Tokutomi, T.; Chida, A.; Asano, Y.; Ishiwata, T.; Koike, Y.; Motegi, A.; Asazuma, T.; Nonoyama, S. A non-obese boy with Prader-Willi syndrome shows cardiopulmonary impairment due to severe kyphoscoliosis. Am. J. Med. Genet. A 2006, 140, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Manzardo, A.M.; Heinemann, J.; Loker, C.; Loker, J. Causes of death in Prader-Willi syndrome: Prader-Willi syndrome association (USA) 40-year mortality survey. Genet. Med. 2017, 19, 635–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grugni, G.; Crino, A.; Bosio, L.; Corrias, A.; Cuttini, M.; De Toni, T.; Di Battista, E.; Franzese, A.; Gargantini, L.; Greggio, N.; et al. The italian national survey for Prader-Willi syndrome: An epidemiologic study. Am. J. Med. Genet. A 2008, 146A, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Lionti, T.; Reid, S.M.; Rowell, M.M. Prader-Willi syndrome in victoria: Mortality and causes of death. J. Paediatr. Child. Health 2012, 48, 506–511. [Google Scholar] [CrossRef]
- Laurance, B.M.; Brito, A.; Wilkinson, J. Prader-Willi syndrome after age 15 years. Arch. Dis. Child. 1981, 56, 181–186. [Google Scholar] [CrossRef]
- Wajchenberg, M.; Astur, N.; Kanas, M.; Martins, D.E. Adolescent idiopathic scoliosis: Current concepts on neurological and muscular etiologies. Scoliosis Spinal Disord. 2016, 11, 4. [Google Scholar] [CrossRef] [Green Version]
- Fendri, K.; Patten, S.A.; Kaufman, G.N.; Zaouter, C.; Parent, S.; Grimard, G.; Edery, P.; Moldovan, F. Microarray expression profiling identifies genes with altered expression in adolescent idiopathic scoliosis. Eur. Spine J. 2013, 22, 1300–1311. [Google Scholar] [CrossRef] [Green Version]
- Baschal, E.E.; Wethey, C.I.; Swindle, K.; Baschal, R.M.; Gowan, K.; Tang, N.L.S.; Avarado, D.M.; Haller, G.E.; Dobbs, M.B.; Taylor, M.R.G.; et al. Exome sequencing identifies rare HSPG2 variant associated with familial idiopathic scoliosis. G3 (Bethesda) 2014, 5, 167–174. [Google Scholar] [CrossRef] [Green Version]
- Baschal, E.E.; Swindle, K.; Justice, C.J.; Baschal, R.M.; Perera, A.; Wethey, C.I.; Poole, A.; Pourquie, O.; Tassy, O.; Miller, N.H. Sequencing of the TBX6 gene in families with familial idiopathic scoliosis. Spine Deform. 2015, 3, 288–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grauers, A.; Wang, J.; Einarsdottir, E.; Simony, A.; Danielsson, A.; Åkesson, K.; Ohlin, A.; Halldin, K.; Grabowski, P.; Tenne, M.; et al. Candidate gene analysis and exome sequencing confirm LBX1 as a susceptibility gene for idiopathic scoliosis. Spine J. 2015, 15, 2239–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogura, Y.; Kou, I.; Miura, S.; Takahashi, A.; Xu, L.; Takeda, K.; Takahashi, Y.; Kono, K.; Kawakami, N.; Uno, K.; et al. A functional SNP in BNC2 is associated with adolescent idiopathic scoliosis. Am. J. Hum. Genet. 2015, 97, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.; Chen, C.; Zhou, T.; Yang, S.; Gao, B.; Zhou, H.; Lian, C.; Wu, Z.; Qiu, X.; Yang, X.; et al. Rare coding variants in MAPK7 predispose to adolescent idiopathic scoliosis. Hum. Mutat. 2017, 38, 1500–1510. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G. Magnesium supplement and the 15q11.2 BP1-BP2 microdeletion (Burnside-Butler) syndrome: A potential treatment? Int. J. Mol. Sci. 2019, 20, 2914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassidy, S.B.; Driscoll, D.J. Prader-Willi syndrome. Eur. J. Hum. Genet. 2009, 17, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Soriano, R.M.; Weisz, I.; Houghton, G.R. Scoliosis in the Prader-Willi syndrome. Spine (Phila PA 1976) 1988, 13, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.H. Growth as a corrective force in the early treatment of progressive infantile scoliosis. J. Bone Joint Surg. Br. 2005, 87, 1237–1247. [Google Scholar] [CrossRef]
- Sanders, J.O.; D’Astous, J.; Fitzgerald, M.; Khoury, J.G.; Kishan, S.; Sturm, P.F. Derotational casting for progressive infantile scoliosis. Pediatr. Orthop. 2009, 29, 581–587. [Google Scholar] [CrossRef] [Green Version]
- Oore, J.; Connell, B.; Yaszay, B.; Samdani, A.; Hilaire, T.S.; Flynn, T.; El-Hawary, R.; Children’s Spine Study, G.; Growing Spine Study, G. Growth friendly surgery and serial cast correction in the treatment of early-onset scoliosis for patients with Prader-Willi syndrome. J. Pediatr. Orthop. 2019, 39, e597–e601. [Google Scholar] [CrossRef]
- Accadbled, F.; Odent, T.; Moine, A.; Chau, E.; Glorion, C.; Diene, G.; de Gauzy, J.S. Complications of scoliosis surgery in Prader-Willi syndrome. Spine (Phila PA 1976) 2008, 33, 394–401. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Nagai, T.; Iida, T.; Ozeki, S.; Nohara, Y. Growth hormone supplement treatment reduces the surgical risk for Prader-Willi syndrome patients. Eur. Spine J. 2012, 21 (Suppl. 4), S483–S491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, J.S.; Lee, S.H.; Seo, S.W.; Koo, K.H.; Jin, D.K. The musculoskeletal manifestations of Prader-Willi syndrome. J. Pediatr. Orthop. 2010, 30, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Sinnema, M.; Maaskant, M.A.; van Schrojenstein Lantman-de Valk, H.M.; van Nieuwpoort, I.C.; Drent, M.L.; Curfs, L.M.; Schrander-Stumpel, C.T. Physical health problems in adults with Prader-Willi syndrome. Am. J. Med. Genet. A 2011, 155A, 2112–2124. [Google Scholar] [CrossRef] [PubMed]
- West, L.A.; Ballock, R.T. High incidence of hip dysplasia but not slipped capital femoral epiphysis in patients with Prader-Willi syndrome. J. Pediatr. Orthop. 2004, 24, 565–567. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, G.; Grugni, G.; Piacente, L.; Delvecchio, M.; Ventura, A.; Giordano, P.; Grano, M.; D’Amato, G.; Laforgia, D.; Crino, A.; et al. Analysis of circulating mediators of bone remodeling in Prader-Willi syndrome. Calcif. Tissue Int. 2018, 102, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.S.; Renfree, S.; Lockwood, D.B.; Karlen, J.; Belthur, M. Syndromic scoliosis: National trends in surgical management and inpatient hospital outcomes: A 12-year analysis. Spine (Phila PA 1976) 2019, 44, 1564–1570. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.J.; Schulz, J.F.; Fornari, E.D.; Wollowick, A.L. Complications associated with surgical repair of syndromic scoliosis. Scoliosis 2015, 10, 14. [Google Scholar] [CrossRef] [Green Version]
- de Baat, P.; van Tankeren, E.; de Lind van Wijngaarden, R.F.; de Klerk, L.W. Sudden proximal spinal dislocation with complete spinal cord injury 1 week after spinal fusion in a child with Prader-Willi syndrome: A case report. Spine (Phila PA 1976) 2011, 36, E1765–E1768. [Google Scholar] [CrossRef]
- Cahill, P.J.; Marvil, S.; Cuddihy, L.; Schutt, C.; Idema, J.; Clements, D.H.; Antonacci, M.D.; Asghar, J.; Samdani, A.F.; Betz, R.R. Autofusion in the immature spine treated with growing rods. Spine (Phila PA 1976) 2010, 35, E1199–E1203. [Google Scholar] [CrossRef]
- Sankar, W.N.; Skaggs, D.L.; Yazici, M.; Johnston, C.E., 2nd; Shah, S.A.; Javidan, P.; Kadakia, R.V.; Day, T.F.; Akbarnia, B.A. Lengthening of dual growing rods and the law of diminishing returns. Spine (Phila PA 1976) 2011, 36, 806–809. [Google Scholar] [CrossRef]
- Joyce, T.J.; Smith, S.L.; Rushton, P.R.P.; Bowey, A.J.; Gibson, M.J. Analysis of explanted magnetically controlled growing rods from seven UK spinal centers. Spine (Phila PA 1976) 2018, 43, E16–E22. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Lee, J.; Cox, D.M.; Manzardo, A.M.; Gold, J.A.; Miller, J.L.; Roof, E.; Dykens, E.; Kimonis, V.; Driscoll, D.J. Growth charts for Prader-Willi syndrome during growth hormone treatment. Clin. Pediatr. (Phila) 2016, 55, 957–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, C.; Essioux, L.; Teinturier, C.; Tauber, M.; Goffin, V.; Bougneres, P. A common polymorphism of the growth hormone receptor is associated with increased responsiveness to growth hormone. Nat. Genet. 2004, 36, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Roberts, J.; Hayes, J.; Tan, X.; Manzardo, A.M. Growth hormone receptor (GHR) gene polymorphism and Prader-Willi syndrome. Am. J. Med. Genet. A 2013, 161A, 1647–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, M.G.; Hossain, W.; Hassan, M.; Manzardo, A.M. Growth hormone receptor (GHR) gene polymorphism and scoliosis in Prader-Willi syndrome. Growth Horm. IGF Res. 2018, 39, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Cowell, C.T.; Dietsch, S. Adverse events during growth hormone therapy. J. Pediatr. Endocrinol. Metab. 1995, 8, 243–252. [Google Scholar] [CrossRef]
- Kim, J.Y.; Rosenfeld, S.R.; Keyak, J.H. Increased prevalence of scoliosis in Turner syndrome. J. Pediatr. Orthop. 2001, 21, 765–766. [Google Scholar] [CrossRef]
- de Lind van Wijngaarden, R.F.; de Klerk, L.W.; Festen, D.A.; Duivenvoorden, H.J.; Otten, B.J.; Hokken-Koelega, A.C. Randomized controlled trial to investigate the effects of growth hormone treatment on scoliosis in children with Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 2009, 94, 1274–1280. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Murakami, N.; Iida, T.; Asano, S.; Ozeki, S.; Nagai, T. Growth hormone treatment for osteoporosis in patients with scoliosis of Prader-Willi syndrome. J. Orthop. Sci. 2014, 19, 877–882. [Google Scholar] [CrossRef]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Bosse, H.J.P.; Butler, M.G. Clinical Observations and Treatment Approaches for Scoliosis in Prader–Willi Syndrome. Genes 2020, 11, 260. https://doi.org/10.3390/genes11030260
van Bosse HJP, Butler MG. Clinical Observations and Treatment Approaches for Scoliosis in Prader–Willi Syndrome. Genes. 2020; 11(3):260. https://doi.org/10.3390/genes11030260
Chicago/Turabian Stylevan Bosse, Harold J.P., and Merlin G. Butler. 2020. "Clinical Observations and Treatment Approaches for Scoliosis in Prader–Willi Syndrome" Genes 11, no. 3: 260. https://doi.org/10.3390/genes11030260
APA Stylevan Bosse, H. J. P., & Butler, M. G. (2020). Clinical Observations and Treatment Approaches for Scoliosis in Prader–Willi Syndrome. Genes, 11(3), 260. https://doi.org/10.3390/genes11030260