Comparative Transcriptomics Reveals Clues for Differences in Pathogenicity between Hysterothylacium aduncum, Anisakis simplex sensu stricto and Anisakis pegreffii


 ,
, 
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Parasite Samples
2.2. RNA Extraction, Quality Check, cDNA Library Construction and RNA-sequencing
2.3. Bioinformatics Analyses of Sequence Data
2.4. Validation of Transcripts by Quantitative Real-Time PCR
3. Results
3.1. Samples and Species Identification
3.2. The Hysterothylacium aduncum, Anisakis simplex sensu stricto and A. pegreffii Transcriptomes
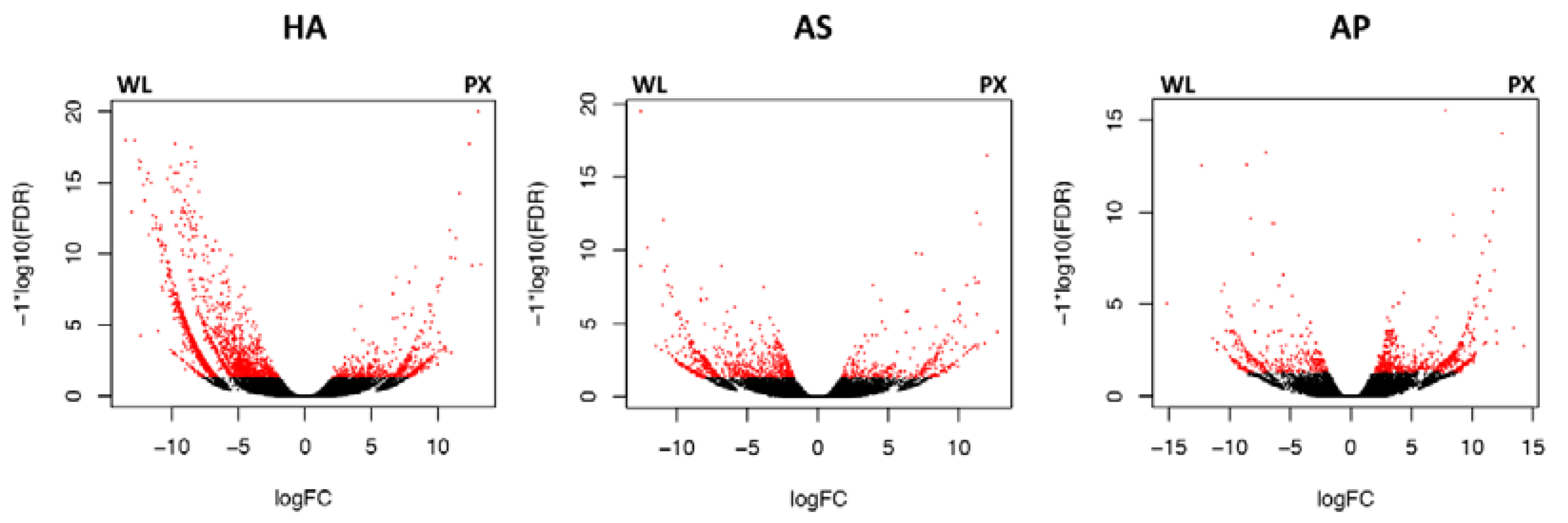
3.3. Differential Gene Expression Analyses of Hysterothylacium aduncum, A. simplex sensu stricto and A. pegreffii
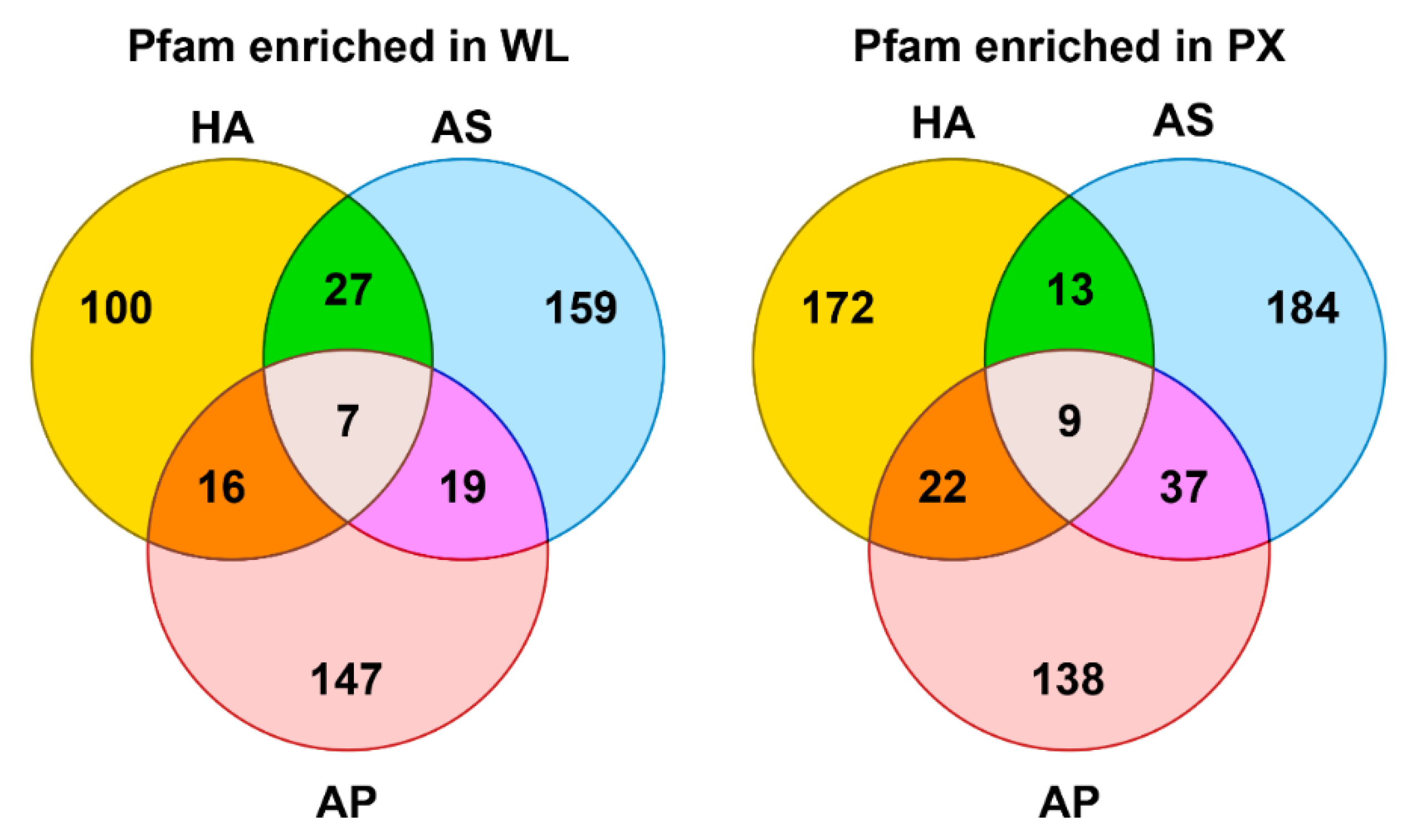
3.4. Transcripts Annotation
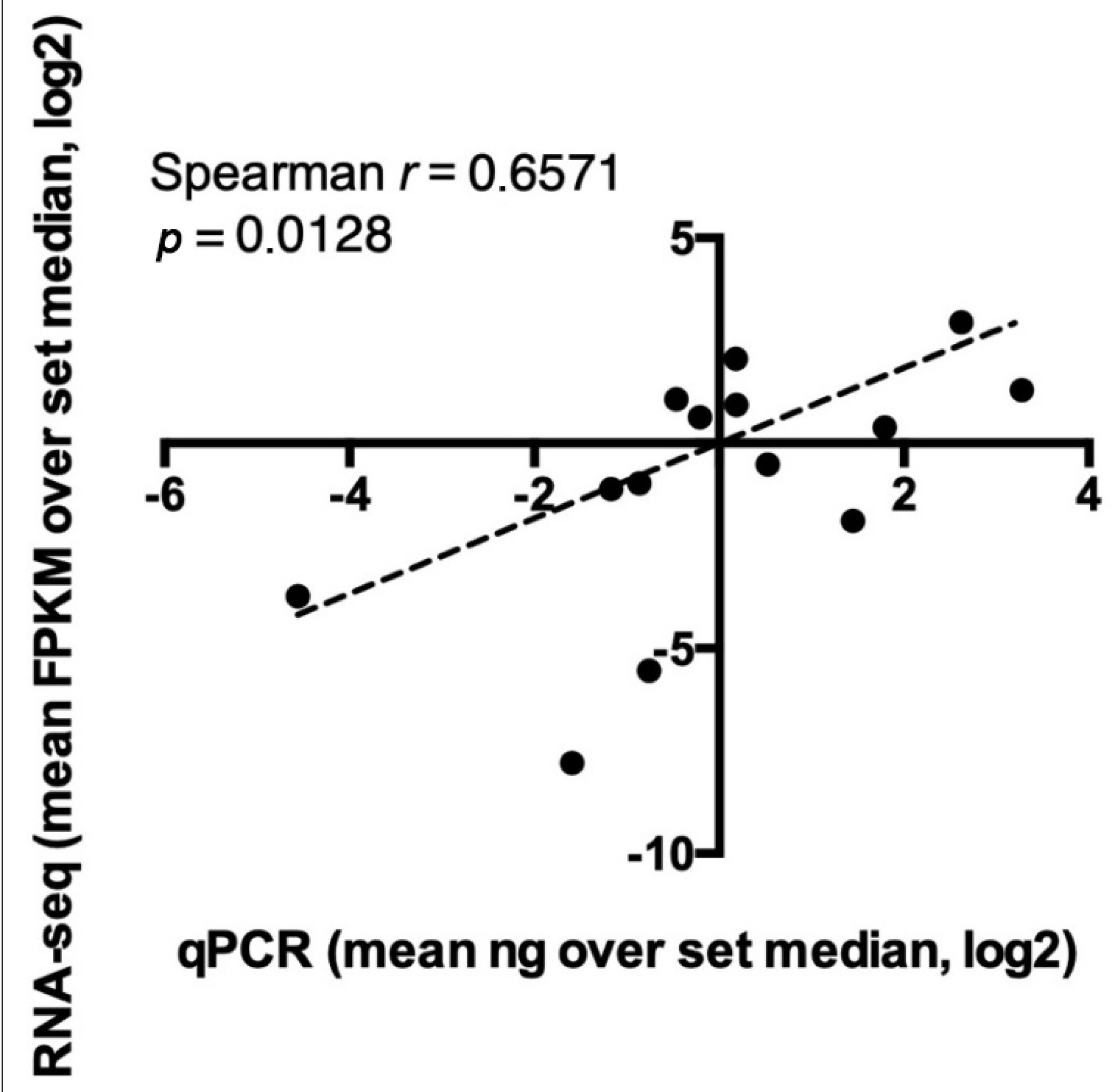
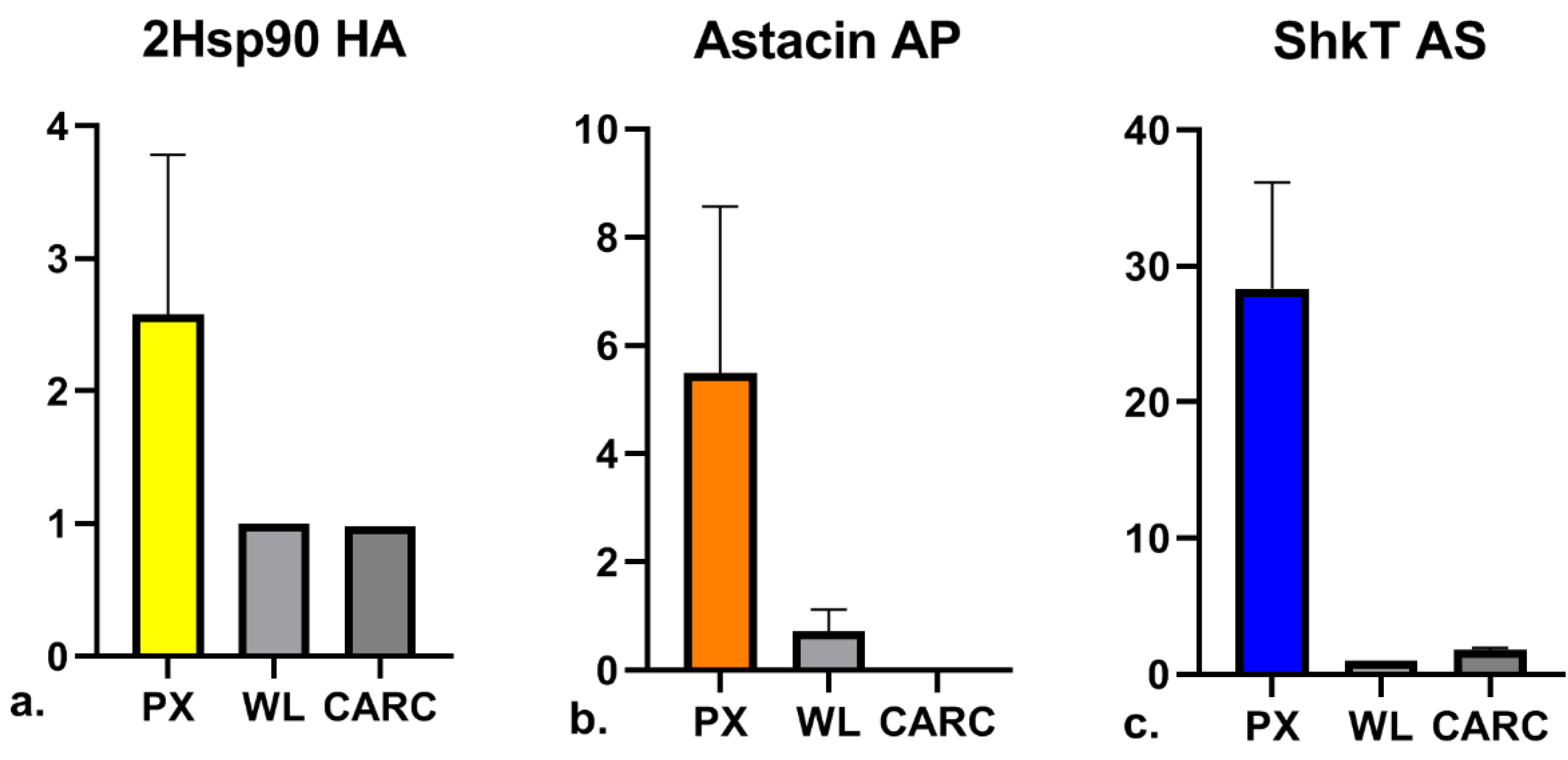
3.5. Relative Quantification of Validated Genes by Real-Time PCR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fagerholm, H.P. Systematic implications of male caudal morphology in ascaridoid nematode parasites. Syst. Parasitol. 1991, 19, 215–228. [Google Scholar] [CrossRef]
- Anderson, R.C. Nematode Parasites of Vertebrates: Their Development and Transmission, 2nd ed.; CABI Publishing: Wallingford, UK, 2000. [Google Scholar]
- Nadler, S.A.; Hudspeth, D.S. Phylogeny of the Ascaridoidea (Nematoda: Ascaridida) based on three genes and morphology: Hypotheses of structural and sequence evolution. J. Parasitol. 2000, 86, 380–393. [Google Scholar] [CrossRef] [Green Version]
- Iglesias, L.; Valero, A.; Gálvez, L.; Benítez, R.; Adroher, F.J. In vitro cultivation of Hysterothylacium aduncum (Nematoda: Anisakidae) from 3rd-stage larvae to egg-laying adults. Parasitology 2002, 125 Pt 5, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Deardorff, T.; Overstreet, R.M. Larval Hysterothylacium (Thynnascaris) (Nematoda: Anisakidae) from fishes and invertebrates in the Gulf of Mexico. Proc. Helminthol. Soc. Wash. 1981, 48, 113–126. [Google Scholar]
- Cipriani, P.; Smaldone, G.; Acerra, V.; D’Angelo, L.; Anastasio, A.; Bellisario, B.; Palma, G.; Nascetti, G.; Mattiucci, S. Genetic identification and distribution of the parasitic larvae of Anisakis pegreffii and Anisakis simplex s.s. in European hake Merluccius merluccius from the Tyrrhenian Sea and Spanish Atlantic coast: Implications for food safety. Int. J. Food Microbiol. 2015, 198, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bao, M.; Pierce, G.; Pascual, S.; González-Muñoz, M.; Mattiucci, S.; Mladineo, I.; Cipriani, P.; Bušelić, I.; Strachan, N.J. Assessing the risk of an emerging zoonosis of worldwide concern: Anisakiasis. Sci. Rep. 2017, 7, 43699. [Google Scholar] [CrossRef] [Green Version]
- Daschner, A.; Cuellar, C.; Rodero, M. The Anisakis allergy debate: Does an evolutionary approach help? Trends Parasitol. 2012, 28, 9–14. [Google Scholar] [CrossRef]
- Nieuwenhuizen, N.E. Anisakis - immunology of a foodborne parasitosis. Parasite Immunol. 2016, 38, 548–557. [Google Scholar] [CrossRef] [Green Version]
- Arizono, N.; Miura, T.; Yamada, M.; Tegoshi, T.; Onishi, K. Human infection with Pseudoterranova azarasi roundworm. Emerg. Infect. Dis. 2011, 17, 555–556. [Google Scholar] [CrossRef]
- Mattiucci, S.; Cipriani, P.; Levsen, A.; Paoletti, M.; Nascetti, G. Molecular Epidemiology of Anisakis and Anisakiasis: An Ecological and Evolutionary Road Map. Adv. Parasitol. 2018, 99, 93–263. [Google Scholar]
- Yagi, K.; Nagasawa, K.; Ishikura, H.; Nakagawa, A.; Sato, N.; Kikuchi, K.; Ishikura, H. Female worm Hysterothylacium aduncum excreted from human: A case report. J. Parasitol. 1996, 45, 12–23. [Google Scholar]
- González-Amores, Y.; Clavijo-Frutos, E.; Salas-Casanova, C.; Alcain-Martínez, G. Direct parasitologial diagnosis of infection with Hysterothylacium aduncum in a patient with epigastralgia. Rev. Esp. Enferm. Dig. 2015, 107, 699–700. [Google Scholar] [PubMed]
- Valero, A.; Terrados, S.; Díaz, V.; Reguera, V.; Lozano, J. Determination of IgE in the serum of patients with allergic reactions to four species of fish-parasite anisakids. J. Investig. Allergol. Clin. Immunol. 2003, 13, 94–98. [Google Scholar] [PubMed]
- Panel EFSA. On biological hazards (BIOHAZ) scientific opinion on risk assessment of parasites in fishery products. EFSA J. 2010, 8, 1543. [Google Scholar] [CrossRef]
- Baird, F.J.; Su, X.; Aibinu, I.; Nolan, M.J.; Sugiyama, H.; Otranto, D.; Lopata, A.L.; Cantacessi, C. The Anisakis Transcriptome Provides a Resource for Fundamental and Applied Studies on Allergy-Causing Parasites. PLoS Negl. Trop. Dis. 2016, 10, e0004845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallero, S.; Lombardo, F.; Su, X.; Salvemini, M.; Cantacessi, C.; D’Amelio, S. Tissue-specific transcriptomes of Anisakis simplex (sensu stricto) and Anisakis pegreffii reveal potential molecular mechanisms involved in pathogenicity. Parasit Vectors 2018, 11, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llorens, C.; Arcos, S.C.; Robertson, L.; Ramos, R.; Futami, R.; Soriano, B.; Ciordia, S.; Careche, M.; González-Muñoz, M.; Jiménez-Ruiz, Y.; et al. Functional insights into the infective larval stage of Anisakis simplex s.s., Anisakis pegreffii and their hybrids based on gene expression patterns. BMC Genom. 2018, 19, 592. [Google Scholar] [CrossRef] [Green Version]
- Hrabar, J.; Trumbić, Ž.; Bočina, I.; Bušelić, I.; Vrbatović, A.; Mladineo, I. Interplay between proinflammatory cytokines, miRNA, and tissue lesions in Anisakis-infected Sprague-Dawley rats. PLoS. Negl. Trop. Dis. 2019, 13, e0007397. [Google Scholar] [CrossRef]
- D’Amelio, S.; Mathiopoulos, K.D.; Santos, C.P.; Pugachev, O.N.; Webb, S.C.; Picanço, M.; Paggi, L. Genetic markers in ribosomal DNA for the identification of members of the genus Anisakis (Nematoda: Ascaridoidea) defined by polymerase-chain-reaction-based restriction fragment length polymorphism. Int. J. Parasitol. 2000, 30, 223–226. [Google Scholar] [CrossRef]
- De Liberato, C.; Bossù, T.; Scaramozzino, P.; Nicolini, G.; Ceddia, P.; Mallozzi, S.; Cavallero, S.; D’Amelio, S. Presence of anisakid larvae in the European anchovy, Engraulis encrasicolus, fished off the Tyrrhenian coast of central Italy. J. Food Prot. 2013, 76, 1643–1648. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WormBase ParaSite. Available online: http://parasite.wormbase.org/Anisakis_simplex_prjeb496/ (accessed on 17 January 2020).
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq: Reference generation and analysis with trinity. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- RSEM (RNA-Seq by Expectation-Maximization). Available online: http://deweylab.biostat.wisc.edu/rsem/ (accessed on 17 January 2020).
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Statist. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Petrella, V.; Aceto, S.; Musacchia, F.; Colonna, V.; Robinson, M.; Benes, V.; Cicotti, G.; Bongiorno, G.; Gradoni, L.; Volf, P.; et al. De novo assembly and sex-specific transcriptome profiling in the sand fly Phlebotomus perniciosus (Diptera, Phlebotominae), a major old world vector of Leishmania infantum. BMC Genom. 2015, 6, 847. [Google Scholar] [CrossRef] [Green Version]
- Musacchia, F.; Basu, S.; Petrosino, G.; Salvemini, M.; Sanges, R. Annocript: A flexible pipeline for the annotation of transcriptomes able to identify putative long noncoding RNAs. Bioinformatics 2015, 31, 2199–2201. [Google Scholar] [CrossRef] [Green Version]
- Aibinu, I.E.; Smooker, P.M.; Lopata, A.L. Anisakis Nematodes in Fish and Shellfish from infection to allergies. Int. J. Parasitol. Parasites Wildl. 2019, 9, 384–393. [Google Scholar] [CrossRef]
- Howe, K.L.; Bolt, B.J.; Shafie, M.; Kersey, P.; Berriman, M. WormBase ParaSite - a comprehensive resource for helminth genomics. Mol. Biochem. Parasitol. 2017, 215, 2–10. [Google Scholar] [CrossRef]
- Zamora, V.; Rodero, M.; Andreu-Ballester, J.C.; Mendez, S.; Cuéllar, C. Induction of tolerogenic properties by Anisakis larval antigens on murine dendritic cells. Parasite Immunol. 2019, 41, e12616. [Google Scholar] [CrossRef]
- Zhou, R.Q.; Ma, G.X.; Korhonen, P.K.; Luo, Y.L.; Zhu, H.H.; Luo, Y.F.; Gasser, R.B.; Xia, Q. Comparative transcriptomic analyses of male and female adult Toxocara canis. Gene. 2017, 600, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Devaney, E. Thermoregulation in the life cycle of nematodes. Int. J. Parasitol. 2006, 36, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuizen, N.E.; Lopata, A.L. Anisakis—A food-borne parasite that triggers allergic host defences. Int. J. Parasitol. 2013, 43, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Asnoussi, A.; Aibinu, I.E.; Gasser, R.B.; Lopata, A.L.; Smooker, P.M. Molecular and immunological characterisation of tropomyosin from Anisakis pegreffii. Parasitol. Res. 2017, 116, 3291–3301. [Google Scholar] [CrossRef] [PubMed]
- González-Fernández, J.; Rivas, L.; Luque-Ortega, J.R.; Núñez-Ramírez, R.; Campioli, P.; Gárate, T.; Perteguer, M.J.; Daschner, A.; Cuéllar, C. Recombinant vs native Anisakis haemoglobin (Ani s 13): Its appraisal as a new gold standard for the diagnosis of allergy. Exp. Parasitol. 2017, 181, 119–129. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HA | AS | AP | AS WB | ||
|---|---|---|---|---|---|
| Total pooled reads | 62,310,036 | 70,465,563 | 59,779,475 | / | |
| Assembly NO FILTER | Total trinity ‘genes’: | 136,550 | 88,087 | 85,068 | 20,971 |
| Total trinity transcripts: | 183,043 | 117,899 | 118,087 | 20,971 | |
| Contig N50: | 613 | 1236 | 1762 | 1197 | |
|
Assembly FILTERED FPKM10 | Total trinity ‘genes’: | 30,254 | 20,574 | 20,840 | 9362 |
| Total trinity transcripts: | 34,603 | 23,469 | 24,120 | 9362 | |
| Contig N50: | 341 | 839 | 1466 | 1218 | |
|
Assembly FILTERED FPKM1 | Total trinity ‘genes’: | 112,416 | 74,738 | 77,406 | 15,811 |
| Total trinity transcripts: | 145,023 | 94,906 | 101,018 | 15,811 | |
| Contig N50: | 504 | 1071 | 1521 | 1281 |
| HA | AS | AP | ||
|---|---|---|---|---|
| Assembly filtered FPKM1 | edgeR included transcripts | 145,023 | 94,906 | 101,018 |
| PX | p < 0.05 | 471 | 612 | 526 |
| p < 0.01 | 185 | 298 | 213 | |
| Contig N50: | 1178 | 2569 | 2366 | |
| WL | p < 0.05 | 1817 | 320 | 282 |
| p < 0.01 | 1107 | 122 | 127 | |
| Contig N50: | 993 | 2783 | 2298 |
| ID | Total | UP | Description | p-Value | adjp | |
|---|---|---|---|---|---|---|
| Pharyngeal enriched | ||||||
| Conserved domain Protein Family | pfam01400 | 63 | 8 | Astacin (Peptidase family M12A) | 3.34 × 10−17 | 1.76 × 10−15 |
| pfam00183 | 111 | 8 | HSP90 protein | 1.11 × 10−9 | 3.51 × 10−8 | |
| pfam04379 | 3 | 3 | DUF525 Protein of unknown function | 3.85 × 10−25 | 6.09 × 10−23 | |
| pfam01776 | 27 | 3 | Ribosomal_L22e | 2.57 × 10−5 | 5.08 × 10−4 | |
| pfam00067 | 33 | 3 | Cytochrome P450 | 1.8 × 10−3 | 2.59 × 10−3 | |
| Whole larva enriched | ||||||
| pfam01484 | 126 | 38 | Nematode cuticle collagen | 3.70 × 10−74 | 8.55 × 10−72 | |
| pfam01391 | 55 | 19 | Collagen triple helix repeat | 2.26 × 10−41 | 1.74 × 10−39 | |
| pfam00092 | 41 | 16 | VWA von Willebrand factor type A | 2.18 × 10−38 | 1.26 × 10−36 | |
| pfam00246 | 32 | 13 | Peptidase_M14 Zinc carboxypeptidase | 5.11 × 10−32 | 2.36 × 10−30 | |
| pfam01674 | 18 | 12 | Lipase_2 | 5.52 × 10−42 | 6.38 × 10−40 | |
| Pharyngeal enriched | ||||||
| Gene ontology Biological process | GO:0006412 | 758 | 20 | translation | 1.49 × 10−6 | 2.47 × 10−5 |
| GO:0006950 | 133 | 8 | response to stress | 2.72 × 10−8 | 6.30 × 10−7 | |
| GO:0007067 | 72 | 4 | mitosis | 6.35 × 10−4 | 6,14 × 10−3 | |
| GO:0007155 | 80 | 4 | cell adhesion | 1.54 × 10−3 | 1.27 × 10−2 | |
| GO:0051301 | 85 | 4 | cell division | 2.47 × 10−3 | 1.91 × 10−2 | |
| Whole larva enriched | ||||||
| GO:0006457 | 397 | 15 | protein folding | 2.84 × 10−4 | 1.30 × 10−2 | |
| GO:0007275 | 82 | 11 | multicellular organismal development | 8.06 × 10−16 | 7.41 × 10−14 | |
| GO:0006950 | 133 | 8 | response to stress | 8.85 × 10−5 | 5.42 × 10−3 | |
| GO:0006979 | 75 | 5 | response to oxidative stress | 1.47 × 10−4 | 1.94 × 10−3 | |
| GO:0007224 | 3 | 4 | smoothened signaling pathway | 1.89 × 10−27 | 3.47 x 10−25 | |
| Pharyngeal enriched | ||||||
| Gene ontology Molecular function | GO:0003735 | 767 | 20 | structural constituent of ribosome | 5.42 × 10−8 | 5.75 × 10−6 |
| GO:0004222 | 253 | 8 | metalloendopeptidase activity | 1.26 × 10−5 | 1.67 × 10−4 | |
| GO:0005506 | 128 | 7 | iron ion binding | 1.09 × 10−7 | 5.80 × 10−6 | |
| GO:0020037 | 192 | 6 | heme binding | 1.46 × 10−3 | 1.50 × 10−2 | |
| GO:0016705 | 56 | 4 | oxidoreductase activity, acting on paired donors | 9.41 × 10−6 | 2.49 × 10−5 | |
| Whole larva enriched | ||||||
| GO:0042302 | 238 | 65 | structural constituent of cuticle | 4.39 × 10−149 | 5.93 × 10−147 | |
| GO:0003700 | 385 | 38 | sequence-specific DNA binding transcription factor | 3.88 × 10−29 | 1.31 × 10−27 | |
| GO:0043565 | 338 | 37 | sequence-specific DNA binding | 2.76 × 10−32 | 1.86 × 10−30 | |
| GO:0005524 | 3496 | 35 | ATP binding | 3.34 × 10−5 | 3.54 × 10−3 | |
| GO:0016787 | 377 | 16 | hydrolase activity | 8.91 × 10−5 | 8.59 × 10−3 | |
| HA | Description | AS | Description | AP | Description | ||
|---|---|---|---|---|---|---|---|
| PX | p < 0.05 | 471 | 612 | 526 | |||
| Annotated transcripts | 172 | 184 | 224 | ||||
| Six most represented pfam | PF01484 PF00238 PF04758 PF04516 PF02738 PF02064 | Collagen Ribosomal protein Ribosomal protein Transcription factor Dehydrogenase Import receptor | PF01400 PF01431 PF00188 PF01204 PF01663 PF01826 | Astacin Peptidase M13 CAP Hydrolase Phosphodiesterase TIL domain | PF00188 PF01400 PF01431 PF01764 PF00014 PF01060 | CAP superfamily Astacin Peptidase M13 Lipase Kunitz BPTI Transthyretin-like | |
| WL | p < 0.05 | 1817 | 282 | 320 | |||
| Annotated transcripts | 347 | 128 | 187 | ||||
| Six most represented pfam | PF01484 PF00092 PF00246 PF01391 PF01674 PF04144 | Cuticole collagen von Willebrand factor Peptidase M14 Collagen Lipase SCAMP family | PF01060 PF01433 PF07857 PF00450 PF00012 PF07690 | Transthyretin-like Peptidase M1 Transmembrane Serine carboxypeptidase Hsp70 Major Facilitator | PF01433 PF00083 PF07690 PF00307 PF00135 PF00046 | Peptidase M1 Sugar transporter Major facilitator EB1 motif Carboxylesterase Homeodomain |
| HA | AS | AP | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ALL | PX_UP | WL_UP | ALL | PX_UP | WL_UP | ALL | PX_UP | WL_UP | |
| >CD | >21,784 | >206 | >485 | >16,645 | >268 | >184 | >14,653 | >240 | >154 |
| >GOBP | >21,274 | >194 | >307 | >15,715 | >154 | >109 | >16,270 | >224 | >215 |
| >GOCC | >22,924 | >185 | >417 | >16,991 | >198 | >138 | >21,925 | >311 | >252 |
| >GOMF | >33,185 | >263 | >593 | >23,141 | >303 | >158 | >16,365 | >300 | >283 |
| >PWL1 | >2828 | >19 | >27 | >1814 | >10 | >11 | >1715 | >21 | >17 |
| >PWL2 | >2845 | >19 | >28 | >1848 | >10 | >11 | >1745 | >20 | >17 |
| PWL3 | 1310 | 4 | 11 | 832 | 5 | 7 | 740 | 7 | 7 |
| RNAseq (FPKM) | RT-qCR (ng) | |||||
|---|---|---|---|---|---|---|
| Species | Symbol | PX | WL | PX | WL | |
| Hysterothylacium aduncum | TRINITY_DN358353_c0_g4_i7 | 2Hsp90 | 130.80 | 0.31 | 1.11 | 0.32 |
| Hysterothylacium aduncum | TRINITY_DN32835_c0_g1_i4 | 1Hsp90 | 285.98 | 18.58 | 1.10 | 2.65 |
| Anisakis pegreffii | TRINITY_DN21666_c0_g2_i1 | CAP | 531.89 | 106.92 | 5.97 | 0.84 |
| Anisakis pegreffii | TRINITY_DN20560_c0_g2_i7 | CRP | 89.58 | 1.48 | 3.36 | 0.57 |
| Anisakis pegreffii | TRINITY_DN29135_c4_g14_i1 | Astacin | 169.56 | 35.03 | 9.41 | 0.53 |
| Anisakis simplex | ASIM_0001574901 | CRS | 143.95 | 31.65 | 0.71 | 0.43 |
| Anisakis simplex | ASIM_0001787701 | ShKT | 48.19 | 5.17 | 1.40 | 0.04 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavallero, S.; Lombardo, F.; Salvemini, M.; Pizzarelli, A.; Cantacessi, C.; D’Amelio, S. Comparative Transcriptomics Reveals Clues for Differences in Pathogenicity between Hysterothylacium aduncum, Anisakis simplex sensu stricto and Anisakis pegreffii. Genes 2020, 11, 321. https://doi.org/10.3390/genes11030321
Cavallero S, Lombardo F, Salvemini M, Pizzarelli A, Cantacessi C, D’Amelio S. Comparative Transcriptomics Reveals Clues for Differences in Pathogenicity between Hysterothylacium aduncum, Anisakis simplex sensu stricto and Anisakis pegreffii. Genes. 2020; 11(3):321. https://doi.org/10.3390/genes11030321
Chicago/Turabian StyleCavallero, Serena, Fabrizio Lombardo, Marco Salvemini, Antonella Pizzarelli, Cinzia Cantacessi, and Stefano D’Amelio. 2020. "Comparative Transcriptomics Reveals Clues for Differences in Pathogenicity between Hysterothylacium aduncum, Anisakis simplex sensu stricto and Anisakis pegreffii" Genes 11, no. 3: 321. https://doi.org/10.3390/genes11030321
APA StyleCavallero, S., Lombardo, F., Salvemini, M., Pizzarelli, A., Cantacessi, C., & D’Amelio, S. (2020). Comparative Transcriptomics Reveals Clues for Differences in Pathogenicity between Hysterothylacium aduncum, Anisakis simplex sensu stricto and Anisakis pegreffii. Genes, 11(3), 321. https://doi.org/10.3390/genes11030321