The Spectrum of Mutations of Homocystinuria in the MENA Region



Abstract
:1. Introduction
2. The most Common CBS Reported Mutations Worldwide
3. The Prevalence of CBS and Associated Mutations in the MENA Region
4. Recent Novel CBS Mutations
5. Clinical Diagnosis and Disease Detection

{kind=link}
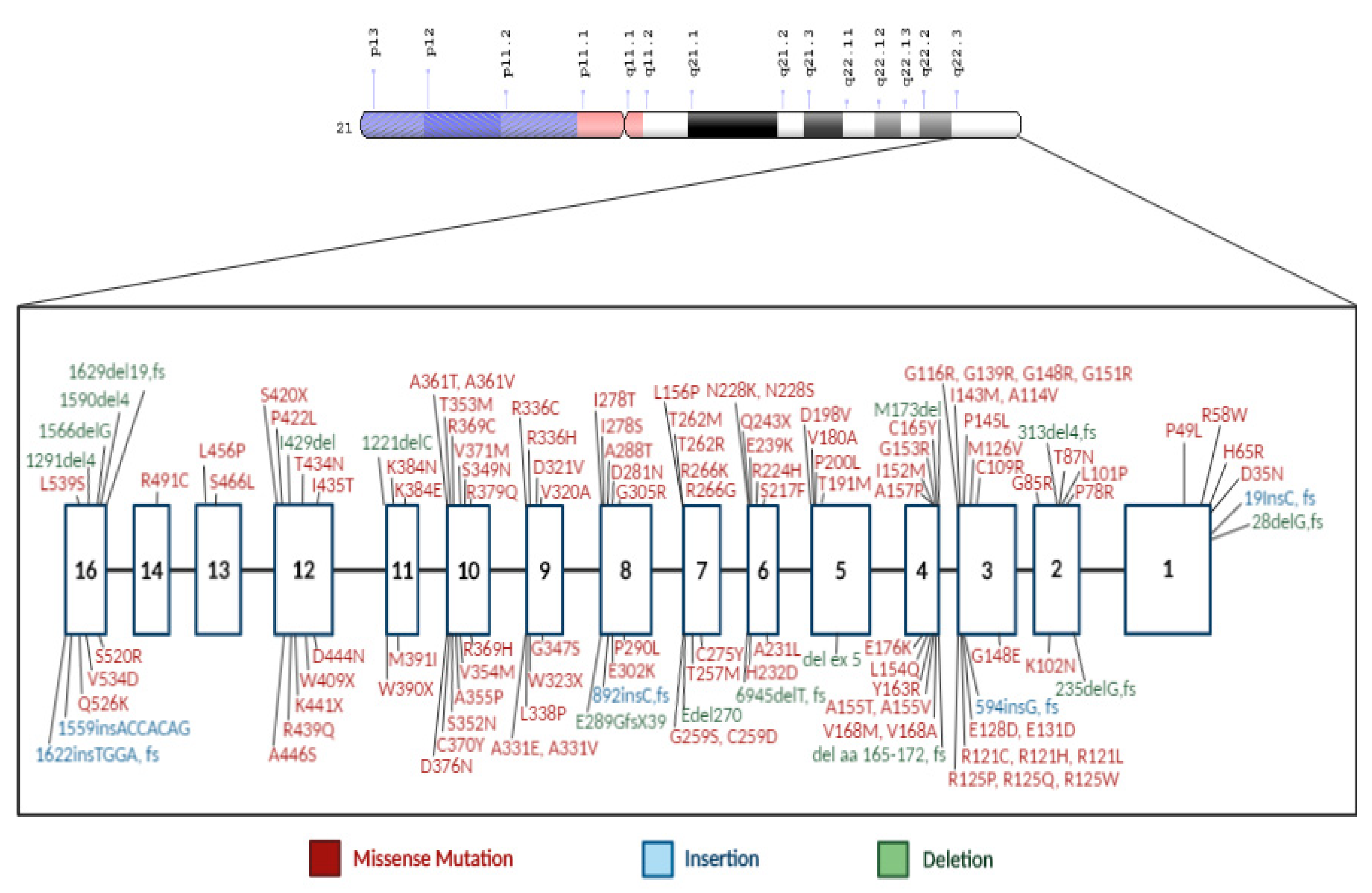{kind=link}
| Country | Nucleotide Change | Protein Change | Exon | Mutation Type | Consanguinity | Pathogenicity and most Common Associated Phenotype Phenotypes | Responsive to Pyridoxine (Vitamin B6) Treatment | Reference |
|---|---|---|---|---|---|---|---|---|
| Qatar | c.1006C > T | p.R336C | 11 | Missense | - | Pathogenic-Hyperhomocystimia, Disproportionate tall stature, Lens luxation, Thromboembolism, Intellectual disability Seizures | Nonresponsiveness | [9,52,59,67] |
| Yes (98.4% of the patients) | [68] | |||||||
| Yes (84% of the patients) | [71] | |||||||
| c.700G>A | p.D234N | 8 | Missense | Yes | Pathogenic-Skeletal and ocular abnormalities | Nonresponsiveness | ||
| c.1039G>A | p.G347S | 9 | Missense | - | - | Not known | ||
| Saudi Arabia | c.969G>A | p.Trp323X | 9 | Novel nonsense mutation | Yes | Pathogenic-a truncation of the CBS protein and results in the complete loss of the CBS enzyme activity. Elevated plasma levels of homocysteine and methionine | Nonresponsiveness | [42,72] |
| c.1006C>T | p.R336C | 11 | Missense | Yes | Pathogenic-Hyperhomocystimia, Disproportionate tall stature, Lens luxation, Thromboembolism, Intellectual disability Seizures | Nonresponsiveness | [72] | |
| c.457G>A | p.G153R | 4 | Missense | Yes | Likely pathogenic | - | [72] | |
| Oman | 844ins68 | - | 8 | Insertion | - | Aneurysmal subarachnoid hemorrhage | - | [73] |
| Lebanon | c.1152 G>C | p.K384N | 11 | Missense | - | - | - | Unpublished data |
| Sudan | c.770C>T | p.T257M | 7 | Missense | Yes | Pathogenic due to complete loss of CBS enzyme activity | Nonresponsiveness | [55,72] |
| Palestinian Arab | c.304A>C | p.K102Q | 2 | Missense | Yes | Likely benign-Ectopia Lentis, Marphanoid features, Thromboembolic episodes | Variable | [64] |
| c.833T>C | p.I278T | 8 | Missense | Pathogenic-Ectopia lentis, Marphanoid feature, mental retardation, idiopathic infertility | Responsive | |||
| c.785C>G | p.T262R | 7 | Missense | Pathogenic-Thromboembolic episodes, mental retardation | - | |||
| g.1627del 19 | addition of non-coded 17 amino acids | 16 | Deletion/ Frameshift | Pathogenic | - | |||
| IVS17 (g18327del 5). | - | 16 | Deletion | Pathogenic-Ectopia lentis, mental retardation | - | |||
| IVS4 (g6643del 29) | 29 bp deletion (exon 5 skipping) | 5 | Deletion | Pathogenic | - | |||
| Pakistan | c.467T>C | p.L156P | 7 | Missense | Yes | Pathogenic-Developmental and Neurological problems. Myopia and lens dislocation | - | [65] |
| c.808_810del | p.E270del | 10 | In-frame deletion | Yes | Pathogenic Developmental and Neurological problems. Glaucoma and subluxated lens. | - |
6. Current and Potential Treatment Approaches
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Applegarth, D.A.; Toone, J.R. Incidence of inborn errors of metabolism in British Columbia, 1969–1996. Pediatrics 2000, 105, e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudd, S.H.; Levy, H.L.; Kraus, J.P. The Online Metabolic and Molecular Bases of Inherited Disease. Scriver CR 2001, 2007–2056. [Google Scholar]
- Kruger, W. Cystathionine β-synthase deficiency in Georgia (USA): Correlation of clinical and biochemical phenotype with genotype. Hum. Mutat. 2003, 22, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.K.C.; Kan, M.Y. Homocysteine-induced endothelial dysfunction. Ann. Nutr. Metab. 2015, 67, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schienle, H.W. Thrombomodulin and ristocetincofactor in homocystinuria: A study in two siblings. Thromb. Res. 1995, 77, 79–86. [Google Scholar] [CrossRef]
- Scriver, C.R. The Metabolic & Molecular Bases of Inherited Disease; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Mudd, S.H. The natural history of homocystinuria due to cystathionine β-synthase deficiency. Am. J. Hum. Genet. 1985, 37, 1. [Google Scholar]
- Majtan, T. Enzyme replacement therapy ameliorates multiple symptoms of murine homocystinuria. Mol. Ther. 2018, 26, 834–844. [Google Scholar] [CrossRef]
- Ismail, H.M. In silico and in vivo models for Qatari-specific classical homocystinuria as basis for development of novel therapies. Hum. Mutat. 2019, 40, 230–240. [Google Scholar] [CrossRef]
- Kery, V.; Poneleit, L.; Kraus, J.P. Trypsin cleavage of human cystathionine β-synthase into an evolutionarily conserved active core: Structural and functional consequences. Arch. Biochem. Biophys. 1998, 355, 222–232. [Google Scholar] [CrossRef]
- Meier, M. Structure of human cystathionine β-synthase: A unique pyridoxal 5′-phosphate-dependent heme protein. EMBO J. 2001, 20, 3910–3916. [Google Scholar] [CrossRef] [Green Version]
- Kraus, J.P. Cystathionine ²-synthase mutations in homocystinuria. Hum. Mutat. 1999, 13, 362. [Google Scholar] [CrossRef]
- Majtan, T. Active cystathionine β-synthase can be expressed in heme-free systems in the presence of metal-substituted porphyrins or a chemical chaperone. J. Biol. Chem. 2008, 283, 34588–34595. [Google Scholar] [CrossRef] [Green Version]
- Ereño-Orbea, J. Structural insight into the molecular mechanism of allosteric activation of human cystathionine β-synthase by S-adenosylmethionine. Proc. Natl. Acad. Sci. USA 2014, 111, E3845–E3852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignoul, S.; Eggermont, J. CBS domains: Structure, function, and pathology in human proteins. Am. J. Physiol. Cell Physiol. 2005, 289, C1369–C1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majtan, T. Potential pharmacological chaperones for cystathionine beta-synthase-deficient homocystinuria. In Targeting Trafficking in Drug Development; Springer: Berlin, Germany, 2017; pp. 345–383. [Google Scholar]
- Shawky, R.M. Profile of genetic disorders prevalent in northeast region of Cairo, Egypt. Egypt. J. Med. Hum. Genet. 2012, 13, 45–62. [Google Scholar] [CrossRef] [Green Version]
- Vyletal, P. Diversity of cystathionine β-synthase haplotypes bearing the most common homocystinuria mutation c. 833T> C: A possible role for gene conversion. Hum. Mutat. 2007, 28, 255–264. [Google Scholar]
- Skovby, F.; Gaustadnes, M.; Mudd, S.H. A revisit to the natural history of homocystinuria due to cystathionine β-synthase deficiency. Mol. Genet. Metab. 2010, 99, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Sokolova, J. Cystathionine beta-synthase deficiency in Central Europe: Discrepancy between biochemical and molecular genetic screening for homocystinuric alleles. Hum. Mutat. 2001, 18, 548–549. [Google Scholar] [CrossRef]
- Janošík, M. Birth prevalence of homocystinuria in Central Europe: Frequency and pathogenicity of mutation c. 1105C> T (p. R369C) in the cystathionine beta-synthase gene. J. Pediatr. 2009, 154, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Kožich, V. Analysis of CBS alleles in Czech and Slovak patients with homocystinuria: Report on three novel mutations E176K, W409X and 1223+ 37 de199. J. Inherit. Metab. Dis. 1997, 20, 363–366. [Google Scholar] [CrossRef]
- Gallagher, P.M. High frequency (71%) of cystathionine β-synthase mutation G307S in Irish homocystinuria patients. Hum. Mutat. 1995, 6, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S. Mouse modeling and structural analysis of the p. G307S mutation in human cystathionine β-synthase (CBS) reveal effects on CBS activity but not stability. J. Biol. Chem. 2018, 293, 13921–13931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinVar. NM_000071.2(CBS):c.919G>A (p.Gly307Ser). 2019. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/117/ (accessed on 1 February 2020).
- Franchis, R.D. Identical genotypes in siblings with different homocystinuric phenotypes: Identification of three mutations in cystathionine β-synthase using an improved bacterial expression system. Hum. Mol. Genet. 1994, 3, 1103–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Franchis, R. Clinical aspects of cystathionine β-synthase deficiency: How wide is the spectrum? Eur. J. Pediatr. 1998, 157, S67–S70. [Google Scholar] [CrossRef] [PubMed]
- de Franchis, R. Four novel mutations in the cystathionine β-synthase gene: Effect of a second linked mutation on the severity of the homocystinuric phenotype. Hum. Mutat. 1999, 13, 453–457. [Google Scholar] [CrossRef]
- Sperandeo, M. Molecular analysis of patients affected by homocystinuria due to cystathionine β-synthase deficiency: Report of a new mutation in exon 8 and a deletion in intron 11. J. Inherit. Metab. Dis. 1995, 18, 211–214. [Google Scholar] [CrossRef]
- Evangelisti, L. Vascular and connective tissue features in 5 Italian patients with homocystinuria. Int. J. Cardiol. 2009, 134, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Urreizti, R. The p. T191M mutation of the CBS gene is highly prevalent among homocystinuric patients from Spain, Portugal and South America. J. Hum. Genet. 2006, 51, 305. [Google Scholar] [CrossRef] [Green Version]
- Cozar, M. Identification and functional analyses of CBS alleles in Spanish and Argentinian homocystinuric patients. Hum. Mutat. 2011, 32, 835–842. [Google Scholar] [CrossRef] [Green Version]
- Casique, L. Characterization of two pathogenic mutations in cystathionine beta-synthase: Different intracellular locations for wild-type and mutant proteins. Gene 2013, 531, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Aral, B. Two novel mutations (K384E and L539S) in the C-terminal moiety of the cystathionine ²-synthase protein in two French pyridoxine-responsive homocystinuria patients. Hum. Mutat. 1997, 9, 81. [Google Scholar] [CrossRef]
- Moat, S.J. The molecular basis of cystathionine β-synthase (CBS) deficiency in UK and US patients with homocystinuria. Hum. Mutat. 2004, 23, 206. [Google Scholar] [CrossRef] [PubMed]
- Carmel, R.; Jacobsen, D.W. Homocysteine in Health and Disease; Cambridge University Press: Cambridge, UK, 2001. [Google Scholar]
- Kluijtmans, L. Defective cystathionine beta-synthase regulation by S-adenosylmethionine in a partially pyridoxine responsive homocystinuria patient. J. Clin. Investig. 1996, 98, 285–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, M.S. Identical Twins with Crouzon Syndrome: Eight-Year Follow-up, Genetic Considerations, and Operative Management. Craniomaxillofacial Trauma Reconstr. 2017, 10, 286–291. [Google Scholar] [CrossRef]
- Tröndle, U. Molecular and Clinical Characterisation of Homocystinuria in Two Austrian Families with Cystathionine β-Synthase Deficiency. Acta Medica Austriaca 2001, 28, 145–151. [Google Scholar] [CrossRef]
- The Human Gene Mutation Database. Cystathionine-Beta-Synthase. 2019. Available online: http://www.hgmd.cf.ac.uk/ac/gene.php?gene=CBS (accessed on 25 January 2020).
- Garland, J. Homocystinuria: Challenges in diagnosis and management. Paediatr. Child Health 1999, 4, 557–562. [Google Scholar] [CrossRef] [Green Version]
- Poloni, S. CBS mutations are good predictors for B6-responsiveness: A study based on the analysis of 35 Brazilian Classical Homocystinuria patients. Mol. Genet. Genom. Med. 2018, 6, 160–170. [Google Scholar] [CrossRef] [Green Version]
- Kelly, P. Stroke in young patients with hyperhomocysteinemia due to cystathionine beta-synthase deficiency. Neurology 2003, 60, 275–279. [Google Scholar] [CrossRef]
- Kožich, V.; Kraus, J.P. Screening for mutations by expressing patient cDNA segments in E. coli: Homocystinuria due to cystathionine β-synthase deficiency. Hum. Mutat. 1992, 1, 113–123. [Google Scholar]
- Sperandeo, M.P. A 68-bp insertion found in a homocystinuric patient is a common variant and is skipped by alternative splicing of the cystathionine beta-synthase mRNA. Am. J. Hum. Genet. 1996, 59, 1391. [Google Scholar]
- Kruger, W.D.; Cox, D.R. A yeast assay for functional detection of mutations in the human cystathionine β-synthase gene. Hum. Mol. Genet. 1995, 4, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Magner, M. Vascular presentation of cystathionine beta-synthase deficiency in adulthood. J. Inherit. Metab. Dis. Off. J. Soc. Study Inborn Errors Metab. 2011, 34, 33–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasim, M. Aminoacidopathies: Prevalence, etiology, screening, and treatment options. Biochem. Genet. 2018, 56, 7–21. [Google Scholar] [CrossRef] [PubMed]
- CAGS. Homocystinuria; Centre for Arab Genomic Studies: Dubai, UAE, 2020. [Google Scholar]
- Moammar, H. Incidence and patterns of inborn errors of metabolism in the Eastern Province of Saudi Arabia, 1983–2008. Ann. Saudi Med. 2010, 30, 271–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, G. Aminoacidopathies among institutionalised mentally retarded in Kuwait. Clin. Genet. 1992, 42, 212. [Google Scholar] [CrossRef] [PubMed]
- Zschocke, J. Molecular neonatal screening for homocystinuria in the Qatari population. Hum. Mutat. 2009, 30, 1021–1022. [Google Scholar] [CrossRef] [PubMed]
- Gaustadnes, M. The molecular basis of cystathionine beta-synthase deficiency in Australian patients: Genotype-phenotype correlations and response to treatment. Hum. Mutat. 2002, 20, 117–126. [Google Scholar] [CrossRef]
- Urreizti, R. Spectrum of CBS mutations in 16 homocystinuric patients from the Iberian Peninsula: High prevalence of T191M and absence of I278T or G307S. Hum. Mutat. 2003, 22, 103. [Google Scholar] [CrossRef]
- Lee, S.J. Identification and functional analysis of cystathionine beta-synthase gene mutations in patients with homocystinuria. J. Hum. Genet. 2005, 50, 648–654. [Google Scholar] [CrossRef]
- Gan-Schreier, H. Newborn population screening for classic homocystinuria by determination of total homocysteine from Guthrie cards. J. Pediatr. 2010, 156, 427–432. [Google Scholar] [CrossRef]
- De Lucca, M.; Casique, L. Characterization of cystathionine beta-synthase gene mutations in homocystinuric Venezuelan patients: Identification of one novel mutation in exon 6. Mol. Genet. Metab. 2004, 81, 209–215. [Google Scholar] [CrossRef] [PubMed]
- El-Said, M.F. A common mutation in the CBS gene explains a high incidence of homocystinuria in the Qatari population. Hum. Mutat. 2006, 27, 719. [Google Scholar] [CrossRef] [PubMed]
- Elsaid, M.F. Are heterocygotes for classical homocystinuria at risk of vitamin B12 and folic acid deficiency? Mol. Genet. Metab. 2007, 92, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Mandava, P. What is the Prevalence of Homocystinuria and Homocysteinemia? 2018. Available online: https://www.medscape.com/answers/1952251–115701/what-is-the-prevalence-of-homocystinuria-and-homocysteinemia (accessed on 10 February 2020).
- Chamberlin, M.E. Methionine adenosyltransferase I/III deficiency: Novel mutations and clinical variations. Am. J. Hum. Genet. 2000, 66, 347–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludolph, A.C. Functional and morphological deficits in late-treated patients with homocystinuria: A clinical, electrophysiologic and MRI study. Acta Neurol. Scand. 1991, 83, 161–165. [Google Scholar] [CrossRef]
- Sarov, M. A case of homocystinuria due to CBS gene mutations revealed by cerebral venous thrombosis. J. Neurol. Sci. 2014, 336, 257–259. [Google Scholar] [CrossRef]
- Gat-Yablonski, G. Homocystinuria in the Arab population of Israel: Identification of two novel mutations using DGGE analysis. Hum. Mutat. 2000, 16, 372. [Google Scholar] [CrossRef]
- Ibrahim, S. CBS mutations and MTFHR SNPs causative of hyperhomocysteinemia in Pakistani children. Mol. Boil. Rep. 2018, 45, 353–360. [Google Scholar] [CrossRef]
- Schiff, M.; Blom, H.J. Treatment of inherited homocystinurias. Neuropediatrics 2012, 43, 295. [Google Scholar]
- Gupta, S. Analysis of the Qatari R336C Cystathionine β-Synthase Protein in Mice. J. Inherit. Metab. Dis. 2019, 42, 831–838. [Google Scholar] [CrossRef]
- Al-Dewik, N. Natural history, with clinical, biochemical, and molecular characterization of classical homocystinuria in the Qatari population. J. Inherit. Metab. Dis. 2019, 42, 813–830. [Google Scholar] [CrossRef] [PubMed]
- Carson, N.A.; Neill, D. Metabolic abnormalities detected in a survey of mentally backward individuals in Northern Ireland. Arch. Dis. Child. 1962, 37, 505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudd, S.H. Homocystinuria: An enzymatic defect. Liver 1964, 1, 1. [Google Scholar] [CrossRef] [PubMed]
- El Bashir, H. Neurodevelopmental and cognitive outcomes of classical homocystinuria: Experience from Qatar. In JIMD Reports; Springer: Berlin, Germany, 2014; Volume 21, pp. 89–95. [Google Scholar]
- Zaidi, S. Clinical and molecular findings of 13 families from Saudi Arabia and a family from Sudan with homocystinuria. Clin. Genet. 2012, 81, 563–570. [Google Scholar] [CrossRef]
- Pathare, A. Hereditary thrombophilia in ethnic Omani patients. Am. J. Hematol. 2006, 81, 101–106. [Google Scholar] [CrossRef]
- Al Essa, M.; Rashed, M.; Ozand, P. Classic homocystinuria: Clinical, biochemical and radiological observations, and therapeutic outcome of 24 Saudi patients. EMHJ East. Mediterr. Health J. 1999, 5, 1196–1203. [Google Scholar]
- Mahmud, I. Decoding the Metabolome and Lipidome of Child. Malnutrition by Mass Spectrometric Techniques: Present Status and Future Perspectives. Anal. Chem. 2019, 91, 14784–14791. [Google Scholar] [CrossRef]
- Homocystinuria. Public Awareness of Homocystinuria. 2014. Available online: https://homocystinuria.wordpress.com/ (accessed on 12 February 2020).
- Longhi, R.C. Cystathionine β-synthase deficiency: A qualitative abnormality of the deficient enzyme modified by vitamin B 6 therapy. Pediatr. Res. 1977, 11, 100. [Google Scholar] [CrossRef] [Green Version]
- Cortez, L.; Sim, V. The therapeutic potential of chemical chaperones in protein folding diseases. Prion 2014, 8, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.-X.; Conn, P.M. Pharmacoperones as novel therapeutics for diverse protein conformational diseases. Physiol. Rev. 2018, 98, 697–725. [Google Scholar] [CrossRef]
- Kopecka, J. Restoring assembly and activity of cystathionine beta-synthase mutants by ligands and chemical chaperones. J. Inherit. Metab. Dis. 2011, 34, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uppala, J.K.; Gani, A.R.; Ramaiah, K.V. Chemical chaperone, TUDCA unlike PBA, mitigates protein aggregation efficiently and resists ER and non-ER stress induced HepG2 cell death. Sci. Rep. 2017, 7, 3831. [Google Scholar] [CrossRef] [PubMed]
- Vang, S. The unexpected uses of urso-and tauroursodeoxycholic acid in the treatment of non-liver diseases. Glob. Adv. Health Med. 2014, 3, 58–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, N.; Narahara, H. Preclinical studies of chemotherapy using histone deacetylase inhibitors in endometrial cancer. Obstet. Gynecol. Int. 2010, 2010, 8. [Google Scholar] [CrossRef] [Green Version]
- Wilcken, D.E.; Dudman, N.P.; Tyrrell, P.A. Homocystinuria due to cystathionine β-synthase deficiency—The effects of betaine treatment in pyridoxine-responsive patients. Metabolism 1985, 34, 1115–1121. [Google Scholar] [CrossRef]
- Bublil, E.M. Enzyme replacement with PEGylated cystathionine beta-synthase ameliorates homocystinuria in murine model. J. Clin. Investig. 2016, 126, 2372–2384. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S. Correction of cystathionine beta-synthase deficiency in mice by treatment with proteasome inhibitors. Hum. Mutat. 2013, 34, 1085–1093. [Google Scholar] [CrossRef] [Green Version]
- Singh, L.R. Activation of mutant enzyme function in vivo by proteasome inhibitors and treatments that induce Hsp70. PLoS Genet. 2010, 6, e1000807. [Google Scholar] [CrossRef] [Green Version]
- Gallego-Villar, L. Cysteamine revisited: Repair of arginine to cysteine mutations. J. Inherit. Metab. Dis. 2017, 40, 555–567. [Google Scholar] [CrossRef]
- Majtan, T.; Pey, A.L.; Kraus, J.P. Kinetic stability of cystathionine beta-synthase can be modulated by structural analogs of S-adenosylmethionine: Potential approach to pharmacological chaperone therapy for homocystinuria. Biochimie 2016, 126, 6–13. [Google Scholar] [CrossRef]
- Pey, A.L. Human cystathionine β-synthase (CBS) contains two classes of binding sites for S-adenosylmethionine (SAM): Complex. regulation of CBS activity and stability by SAM. Biochem. J. 2013, 449, 109–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L. Modulation of cystathionine β-synthase level regulates total serum homocysteine in mice. Circ. Res. 2004, 94, 1318–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-O. Treatment of Cystathionine β-Synthase Deficiency in Mice Using a Minicircle-Based Naked DNA Vector. Hum. Gene Ther. 2019, 30, 1093–1100. [Google Scholar] [CrossRef] [PubMed]


| Country | Nucleotide Change | Protein Change | Exon | Mutation Type | Consanguinity | Pathogenicity and most Common Associated Phenotype Phenotypes | Responsive to Pyridoxine (Vitamin B6) Treatment | Reference |
|---|---|---|---|---|---|---|---|---|
| Western Eurasians | c.833T>C | p.I278T | 8 | Missense | No | Pathogenic-Ectopia lentis, Marphanoid feature, mental retardation, idiopathic infertility, severe vascular complications | Responsiveness | [18] |
| Denmark Norway Germany Czech and Slovak Republics | c.833T>C | p.I278T | 8 | Missense | - | Pathogenic-Ectopia lentis, Marphanoid feature, mental retardation, idiopathic infertility, severe vascular complications | Responsiveness | [19,20] |
| Czech | c.1105C>T | p.R369C | 10 | Missense | - | Patients do not have any clinical symptoms at all. The late onset of symptoms, lack of mental retardation, and connective tissue involvement | Responsiveness | [21] |
| Slovakia | 526 G>A | p.E176K | 4 | Missense | No | Conflicting interpretations of pathogenicity. Thoracic aortic aneurysm and aortic dissection. Cardiovascular phenotype | Nonresponsiveness | [22] |
| 1226 G>A | p.W409X | 12 | Nonsense | No | - | Nonresponsiveness | ||
| Netherland | c.833T>C | p.I278T | 8 | Missense | No | Pathogenic-Ectopia lentis, Marphanoid feature, mental retardation, idiopathic infertility, severe vascular complications | Responsiveness | [12,19] |
| c.539 T>C | p.V180A | 5 | Missense | No | - | - | [12] | |
| c.1330G>A | p.D444N | 12 | Missense | No | Mild | - | ||
| Ireland | c.919 G>A | p.G307S | 8 | Missense | - | Pathogenic-Cardiovascular phenotype | Nonresponsiveness | [23,24,25] |
| c.306 G>C | p.K102N | 2 | Missense | No | Likely pathogenic-Neurogenic bladder or bowel signs, developmental delay, mental retardation | Responsiveness | [26] | |
| Italy | c.146 C>T | p.P49L | 1 | Missense | No | Pathogenic, VUS Cardiovascular phenotype | Responsiveness | [27] |
| c.172 C>T | p.R58W | 1 | Missense | No | Mild mental retardation with EEG anomalies, osteoporosis, malar flush, and ultrasound evidence of arterial disease | Nonresponsiveness | [28] | |
| c.262 C>T | p.P88S | 2 | Missense | - | Pathogenic-Zonular pulverulent cataract phenotype | - | [29] | |
| c.469G<C | p.A157P | 4 | Missense | No | Pathogenic-Pectus escavatum, Tricuspid valve prolapse, Kyphoscoliosis, Iridodonesis, Mild elbow valgus | - | [30] | |
| Spain | c.869 C>T | p.P290L | 8 | Missense | No | Hermansky Pudlak syndrome 2 | Responsiveness | [29] |
| c.572C>T | p.T191M | 5 | Missense | No | Pathogenic | Nonresponsiveness | [31] | |
| c.833T<G | p.I278S | 8 | Missense | No | Pathogenic | Responsiveness | [32] | |
| Portugal | c.572C>T | p.T191M | 5 | Missense | No | Pathogenic | Nonresponsiveness | [31] |
| Venezuela | c.700G>A | p.D234N | 8 | Missense | Yes | Pathogenic-Skeletal and ocular abnormalities | Nonresponsiveness | [33] |
| France | 1150 A>G | p.K384E | 11 | Missense | No | Pathogenic | Responsiveness | [34] |
| 1616 T>C | p.L539S | 16 | Missense | No | Pathogenic | Responsiveness | ||
| UK | c.374G>A | p.R125Q | 3 | Missense | No | Pathogenic Cardiovascular phenotype | - | [35] |
| c.430G>A | p.E144K | 3 | Missense | No | Pathogenic Cardiovascular phenotype | - | [35] | |
| c.833T>C | p.I278T | 8 | Missense | No | Pathogenic-Ectopia lentis, Marphanoid feature, mental retardation, idiopathic infertility, severe vascular complications | Responsiveness | [35] | |
| c.919G>A | p.G307S | 8 | Missense | No | Pathogenic-Cardiovascular phenotype | Nonresponsiveness | [25,35] | |
| USA | c.341C>T | p.A114V | 3 | Missense | No | - | - | [35] |
| c.374G>A | p.R125Q | 3 | Missense | No | Pathogenic-Cardiovascular phenotype | Nonresponsiveness | [35,36] | |
| c.785C>T | p.T262M | 7 | Missense | No | Pathogenic-Thromboembolic episodes, mental retardation | Nonresponsiveness | [35,36] | |
| c.797G>A | p.R266K | 7 | Missense | No | - | Responsiveness | [35] | |
| c.833T>C | p.I278T | 8 | Missense | No | Pathogenic-Severe vascular complications | Responsiveness | [35] | |
| c.919G>A | p.G307S | 8 | Missense | No | Pathogenic-Cardiovascular phenotype | Nonresponsiveness | [25,35] | |
| g.13217A>C | (del ex 12) | Intron 11 | Deletion | No | - | - | [35] | |
| c.1330G>A | p.D444N | 12 | Missense | No | Pathogenic-Psychomotoric retardation and marfanoid features | Partially pyridoxine-responsive | [35,37] | |
| India | c.518delTGA | p.M173del | 4 | Deletion | No | Pathogenic | - | [32] |
| Argentina | c.676G<A | p.A226T | 6 | Missense | No | Mild-Hypertrophic cardiomyopathy | Responsiveness | [25,32] |
| c.962A<T | p.D321V | 9 | Missense | No | Pathogenic-Crouzon syndrome | - | [32,38] | |
| c.1336G<T | p. A446S | 12 | Missense | No | Mild-lens dislocation | Responsiveness | [32] | |
| Australia | c.833T>C | p.I278T | 8 | Missense | No | Pathogenic-Ectopia lentis, Marphanoid feature, mental retardation, idiopathic infertility, severe vascular complications | Responsiveness | [39] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Sadeq, D.W.; Nasrallah, G.K. The Spectrum of Mutations of Homocystinuria in the MENA Region. Genes 2020, 11, 330. https://doi.org/10.3390/genes11030330
Al-Sadeq DW, Nasrallah GK. The Spectrum of Mutations of Homocystinuria in the MENA Region. Genes. 2020; 11(3):330. https://doi.org/10.3390/genes11030330
Chicago/Turabian StyleAl-Sadeq, Duaa W., and Gheyath K. Nasrallah. 2020. "The Spectrum of Mutations of Homocystinuria in the MENA Region" Genes 11, no. 3: 330. https://doi.org/10.3390/genes11030330
APA StyleAl-Sadeq, D. W., & Nasrallah, G. K. (2020). The Spectrum of Mutations of Homocystinuria in the MENA Region. Genes, 11(3), 330. https://doi.org/10.3390/genes11030330