Increasing the Genetic Diagnosis Yield in Inherited Retinal Dystrophies: Assigning Pathogenicity to Novel Non-canonical Splice Site Variants

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
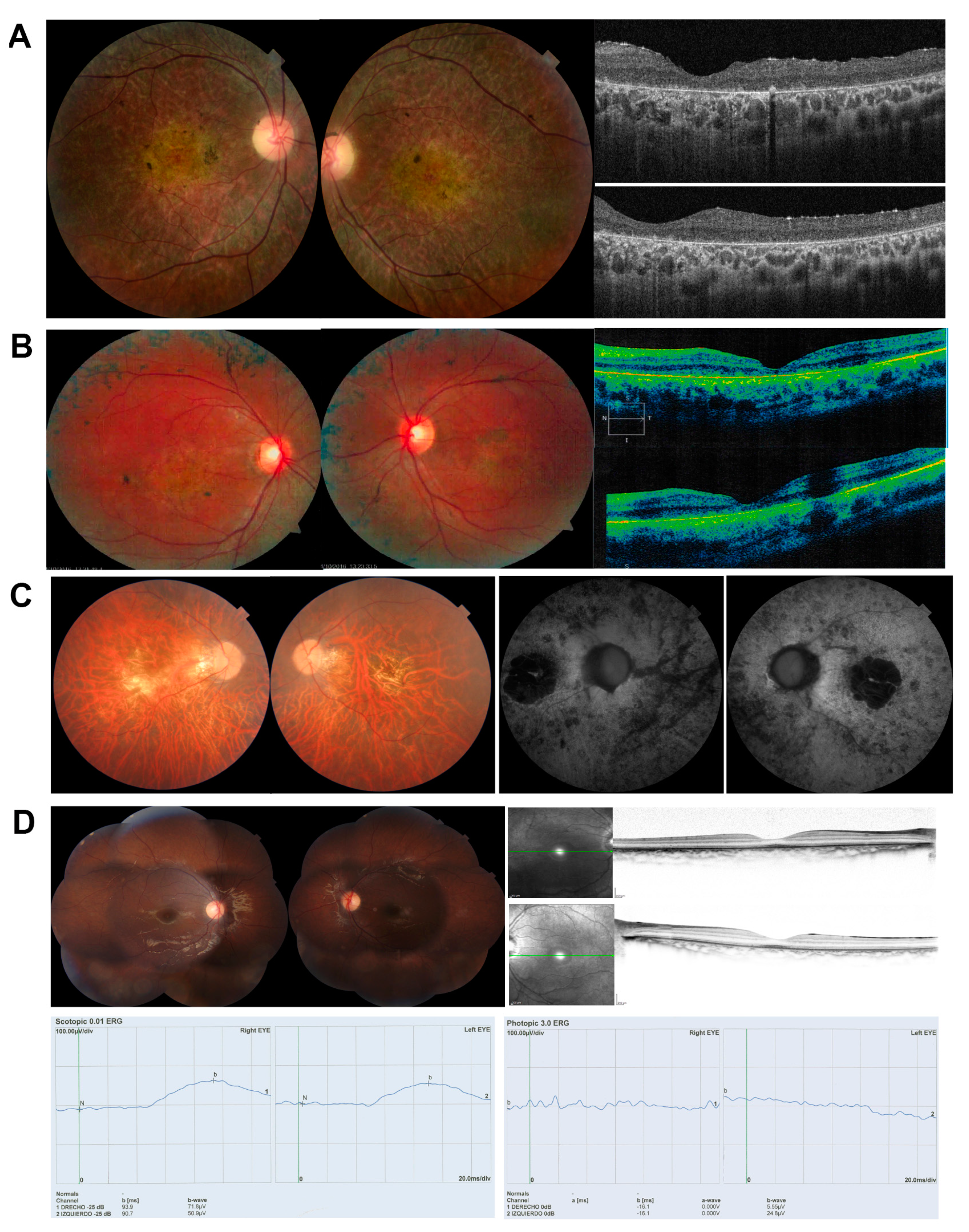
2.1. Clinical Diagnosis
2.2. Samples
2.3. In Silico Analysis of the Variant Effects on Splicing
2.4. In Vivo Splicing Analysis of RP2
2.5. In Vitro Splicing Assays of ABCA4 and POC1B in HEK293T Cells
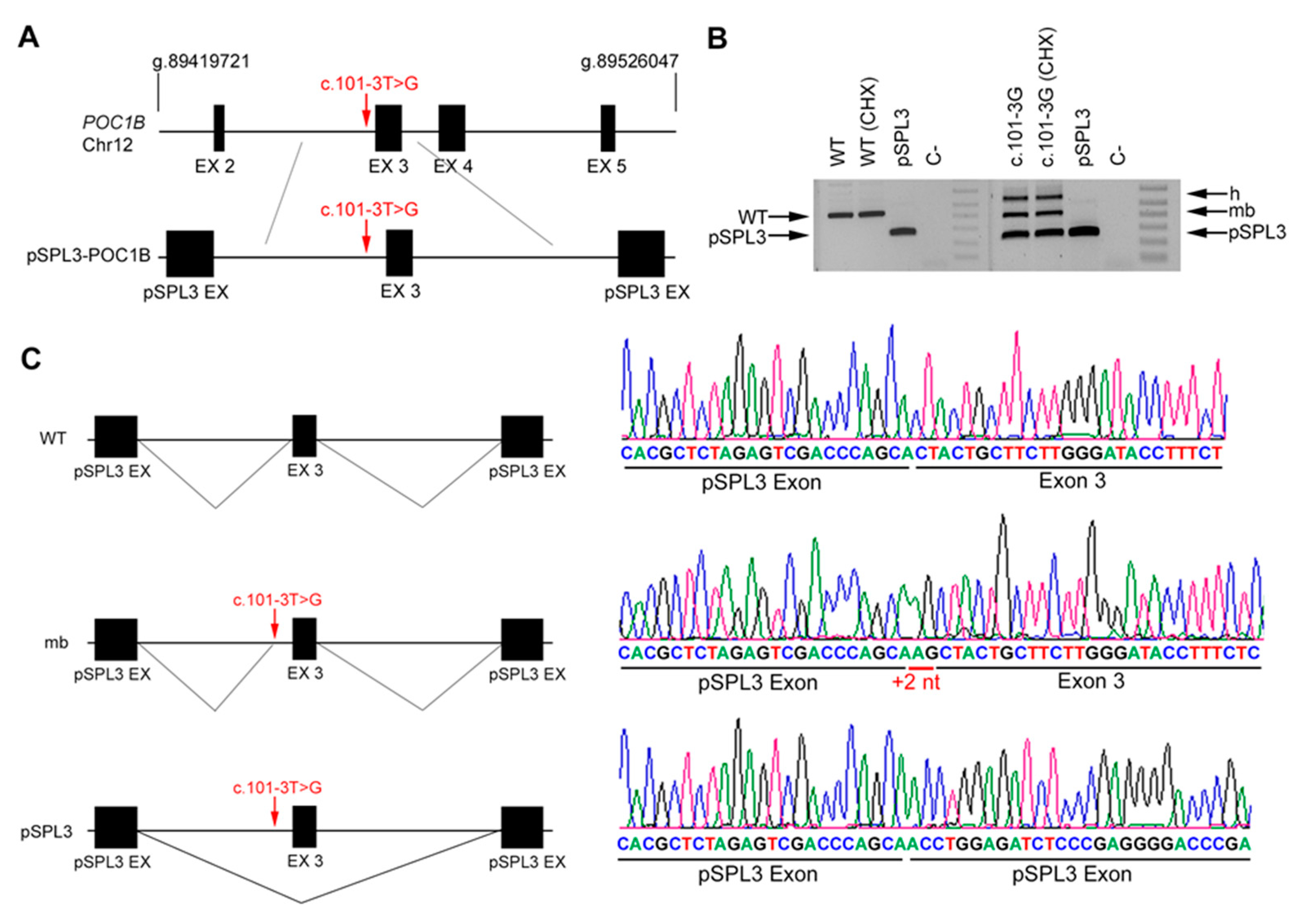
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ellingford, J.M.; Barton, S.; Bhaskar, S.; O’Sullivan, J.; Williams, S.G.; Lamb, J.A.; Panda, B.; Sergouniotis, P.I.; Gillespie, R.L.; Daiger, S.P.; et al. Molecular findings from 537 individuals with inherited retinal disease. J. Med. Genet. 2016, 53, 761–767. [Google Scholar] [CrossRef] [Green Version]
- Zernant, J.; Xie, Y.A.; Ayuso, C.; Riveiro-Alvarez, R.; Lopez-Martinez, M.-A.; Simonelli, F.; Testa, F.; Gorin, M.B.; Strom, S.P.; Bertelsen, M.; et al. Analysis of the ABCA4 genomic locus in Stargardt disease. Hum. Mol. Genet. 2014, 23, 6797–6806. [Google Scholar] [CrossRef] [PubMed]
- Sangermano, R.; Khan, M.; Cornelis, S.S.; Richelle, V.; Albert, S.; Garanto, A.; Elmelik, D.; Qamar, R.; Lugtenberg, D.; van den Born, L.I.; et al. ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease. Genome Res. 2018, 28, 100–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simunovic, M.P.; Jolly, J.K.; Xue, K.; Edwards, T.L.; Groppe, M.; Downes, S.M.; MacLaren, R.E. The spectrum of CHM gene mutations in choroideremia and their relationship to clinical phenotype. Investig. Opthalmol. Vis. Sci. 2016, 57, 6033–6039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibley, C.R.; Blazquez, L.; Ule, J. Lessons from non-canonical splicing. Nat. Rev. Genet. 2016, 17, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, N. Modulation of aberrant splicing in human RNA diseases by chemical compounds. Hum. Genet. 2017, 136, 1237–1245. [Google Scholar] [CrossRef]
- Zhu, Y.; Deng, H.; Chen, X.; Li, H.; Yang, C.; Li, S.; Pan, X.; Tian, S.; Feng, S.; Tan, X.; et al. Skipping of an exon with a nonsense mutation in the DMD gene is induced by the conversion of a splicing enhancer to a splicing silencer. Hum. Genet. 2019, 138, 771–785. [Google Scholar] [CrossRef]
- Valero, R.; de Castro-Miró, M.; Jiménez-Ochoa, S.; Rodríguez-Ezcurra, J.J.; Marfany, G.; Gonzàlez-Duarte, R. Aberrant splicing events associated to CDH23 noncanonical splice site mutations in a proband with atypical Usher syndrome 1. Genes 2019, 10, 732. [Google Scholar] [CrossRef] [Green Version]
- Liquori, A.; Vaché, C.; Baux, D.; Blanchet, C.; Hamel, C.; Malcolm, S.; Koenig, M.; Claustres, M.; Roux, A.-F. Whole USH2A gene sequencing identifies several new deep intronic mutations. Hum. Mutat. 2016, 37, 184–193. [Google Scholar] [CrossRef]
- Jaijo, T.; Aller, E.; Aparisi, M.; García-García, G.; Hernan, I.; Gamundi, M.; Nájera, C.; Carballo, M.; Millán, J. Functional analysis of splicing mutations in MYO7A and USH2A genes. Clin. Genet. 2011, 79, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Pomares, E.; Riera, M.; Castro-Navarro, J.; Andrés-Gutiérrez, Á.; Gonzàlez-Duarte, R.; Marfany, G. Identification of an intronic single-point mutation in RP2 as the cause of semidominant X-linked retinitis pigmentosa. Investig. Opthalmol. Vis. Sci. 2009, 50, 5107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zernant, J.; Lee, W.; Nagasaki, T.; Collison, F.T.; Fishman, G.A.; Bertelsen, M.; Rosenberg, T.; Gouras, P.; Tsang, S.H.; Allikmets, R. Extremely hypomorphic and severe deep intronic variants in the ABCA4 locus result in varying Stargardt disease phenotypes. Mol. Case Stud. 2018, 4, a002733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaz-Drago, R.; Custódio, N.; Carmo-Fonseca, M. Deep intronic mutations and human disease. Hum. Genet. 2017, 136, 1093–1111. [Google Scholar] [CrossRef] [PubMed]
- Lord, J.; Gallone, G.; Short, P.J.; McRae, J.F.; Ironfield, H.; Wynn, E.H.; Gerety, S.S.; He, L.; Kerr, B.; Johnson, D.S.; et al. Pathogenicity and selective constraint on variation near splice sites. Genome Res. 2019, 29, 159–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runhart, E.H.; Valkenburg, D.; Cornelis, S.S.; Khan, M.; Sangermano, R.; Albert, S.; Bax, N.M.; Astuti, G.D.N.; Gilissen, C.; Pott, J.-W.R.; et al. Late-onset Stargardt disease due to mild, deep-intronic ABCA4 alleles. Investig. Opthalmol. Vis. Sci. 2019, 60, 4249. [Google Scholar] [CrossRef] [Green Version]
- Nassisi, M.; Mohand-Saïd, S.; Andrieu, C.; Antonio, A.; Condroyer, C.; Méjécase, C.; Varin, J.; Wohlschlegel, J.; Dhaenens, C.-M.; Sahel, J.-A.; et al. Prevalence of ABCA4 deep-intronic variants and related phenotype in an unsolved “one-hit” cohort with Stargardt disease. Int. J. Mol. Sci. 2019, 20, 5053. [Google Scholar] [CrossRef] [Green Version]
- Steele-Stallard, H.B.; Le Quesne Stabej, P.; Lenassi, E.; Luxon, L.M.; Claustres, M.; Roux, A.-F.; Webster, A.R.; Bitner-Glindzicz, M. Screening for duplications, deletions and a common intronic mutation detects 35% of second mutations in patients with USH2A monoallelic mutations on Sanger sequencing. Orphanet J. Rare Dis. 2013, 8, 122. [Google Scholar] [CrossRef] [Green Version]
- Vervoort, R.; Lennon, A.; Bird, A.C.; Tulloch, B.; Axton, R.; Miano, M.G.; Meindl, A.; Meitinger, T.; Ciccodicola, A.; Wright, A.F. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat. Genet. 2000, 25, 462–466. [Google Scholar] [CrossRef]
- Garanto, A.; Duijkers, L.; Tomkiewicz, T.Z.; Collin, R.W.J. Antisense oligonucleotide screening to optimize the rescue of the splicing defect caused by the recurrent deep-intronic ABCA4 variant c.4539+2001G>A in Stargardt Disease. Genes 2019, 10, 452. [Google Scholar] [CrossRef] [Green Version]
- Fadaie, Z.; Khan, M.; Del Pozo-Valero, M.; Cornelis, S.S.; Ayuso, C.; Cremers, F.P.M.; Roosing, S.; The ABCA Study Group. Identification of splice defects due to noncanonical splice site or deep-intronic variants in ABCA4. Hum. Mutat. 2019, 40, 2365–2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauwens, M.; Garanto, A.; Sangermano, R.; Naessens, S.; Weisschuh, N.; De Zaeytijd, J.; Khan, M.; Sadler, F.; Balikova, I.; Van Cauwenbergh, C.; et al. ABCA4-associated disease as a model for missing heritability in autosomal recessive disorders: Novel noncoding splice, cis-regulatory, structural, and recurrent hypomorphic variants. Genet. Med. 2019, 21, 1761–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangermano, R.; Garanto, A.; Khan, M.; Runhart, E.H.; Bauwens, M.; Bax, N.M.; van den Born, L.I.; Khan, M.I.; Cornelis, S.S.; Verheij, J.B.G.M.; et al. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet. Med. 2019, 21, 1751–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garanto, A.; Collin, R.W.J. Design and in vitro use of antisense oligonucleotides to correct pre-mRNA splicing defects in inherited retinal dystrophies. Methods Mol. Biol. 2018, 1715, 61–78. [Google Scholar]
- Vázquez-Domínguez, I.; Garanto, A.; Collin, R.W.J. Molecular therapies for inherited retinal diseases—Current standing, opportunities and challenges. Genes 2019, 10, 654. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Disease | Age | A.O. | Sex | VA | BCVA | ERG |
|---|---|---|---|---|---|---|---|
| years | years | F/M | RE/LE | RE/LE | |||
| DBG1 | RP | 31 | 12 | F | CF OU (counting fingers) | CF OU | Abolished photopic and scotopic. |
| DBG2 | RP + macula | 22 | 7 | M | 20–320/ 20–200 | 20–200 | No data |
| DBG3 | RP | 40 | 15 | M | Counting fingers | 20–400 | Abolished rod ERG and 88% reduced ERG in cones. |
| DBG4 | CRD | 8 | 3 | M | OU: 20-100 | OD: 20–40 OS: 20–50 | Reduced photopic and flicker amplitude. |
| Allele 1 | Allele 2 | |||||
|---|---|---|---|---|---|---|
| Patient ID | Gene | Exon/Intron | Nucleotide Change | Exon/Intron | Nucleotide Change | |
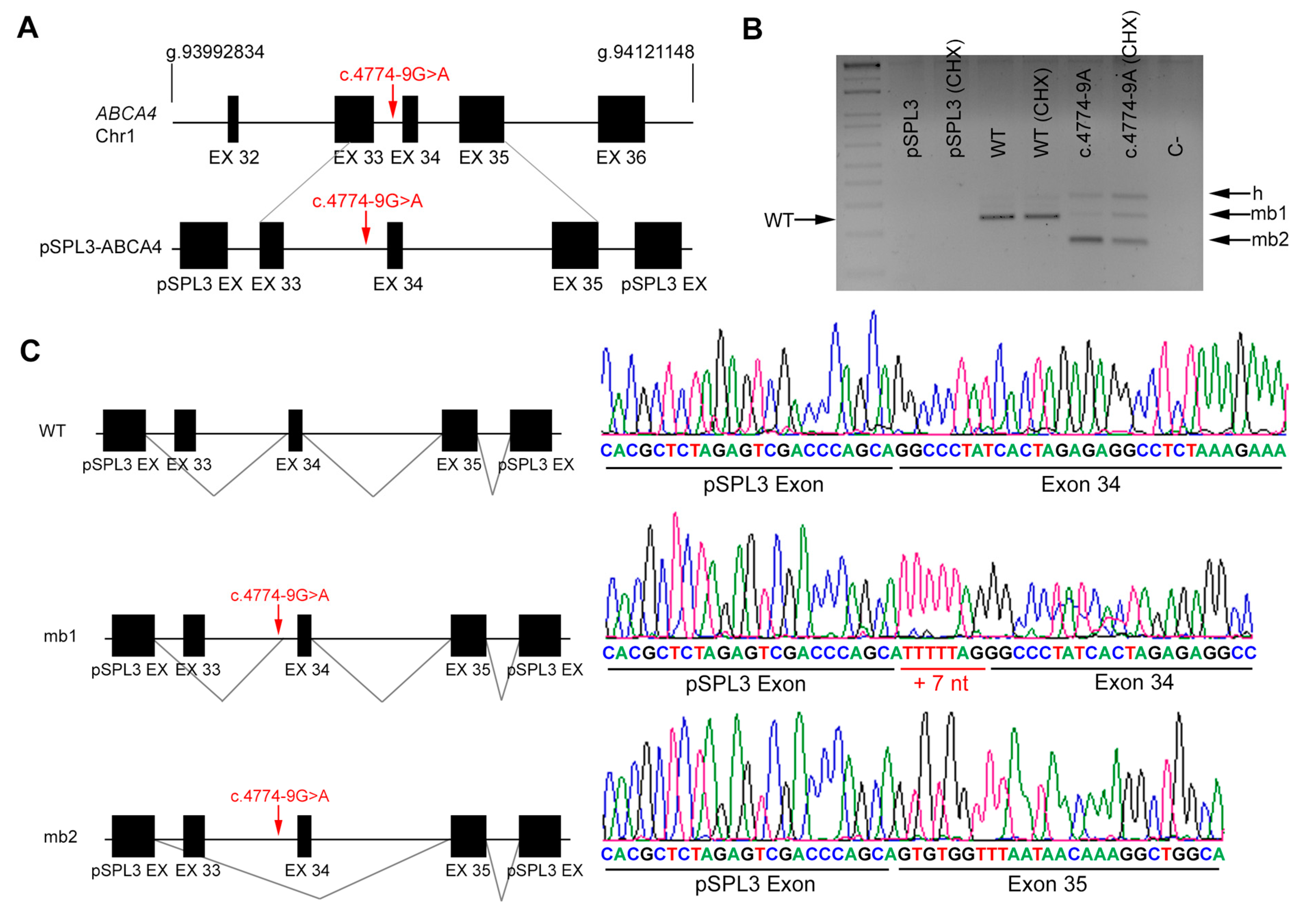
| DBG1 | Index | ABCA4 | 6 | c.735T>G | IVS33 | c.4774-9 G>A |
| Affected sister | c.735T>G | c.4774-9 G>A | ||||
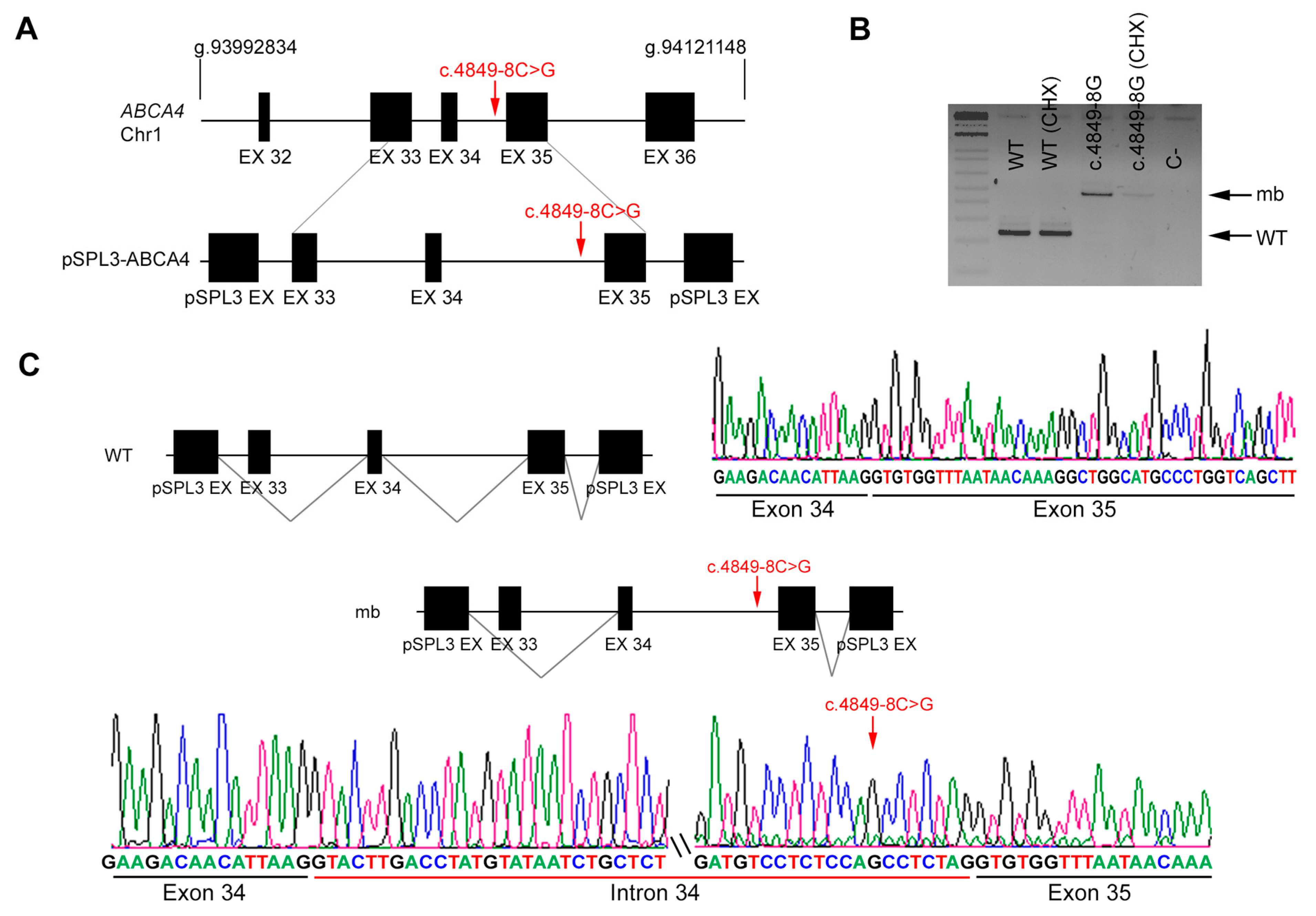
| DBG2 | Index | ABCA4 | 19 | c.2894A>G | IVS34 | c.4849-8 C>G |
| Unaffected mother | Reference sequence (WT) | c.4849-8 C>G | ||||
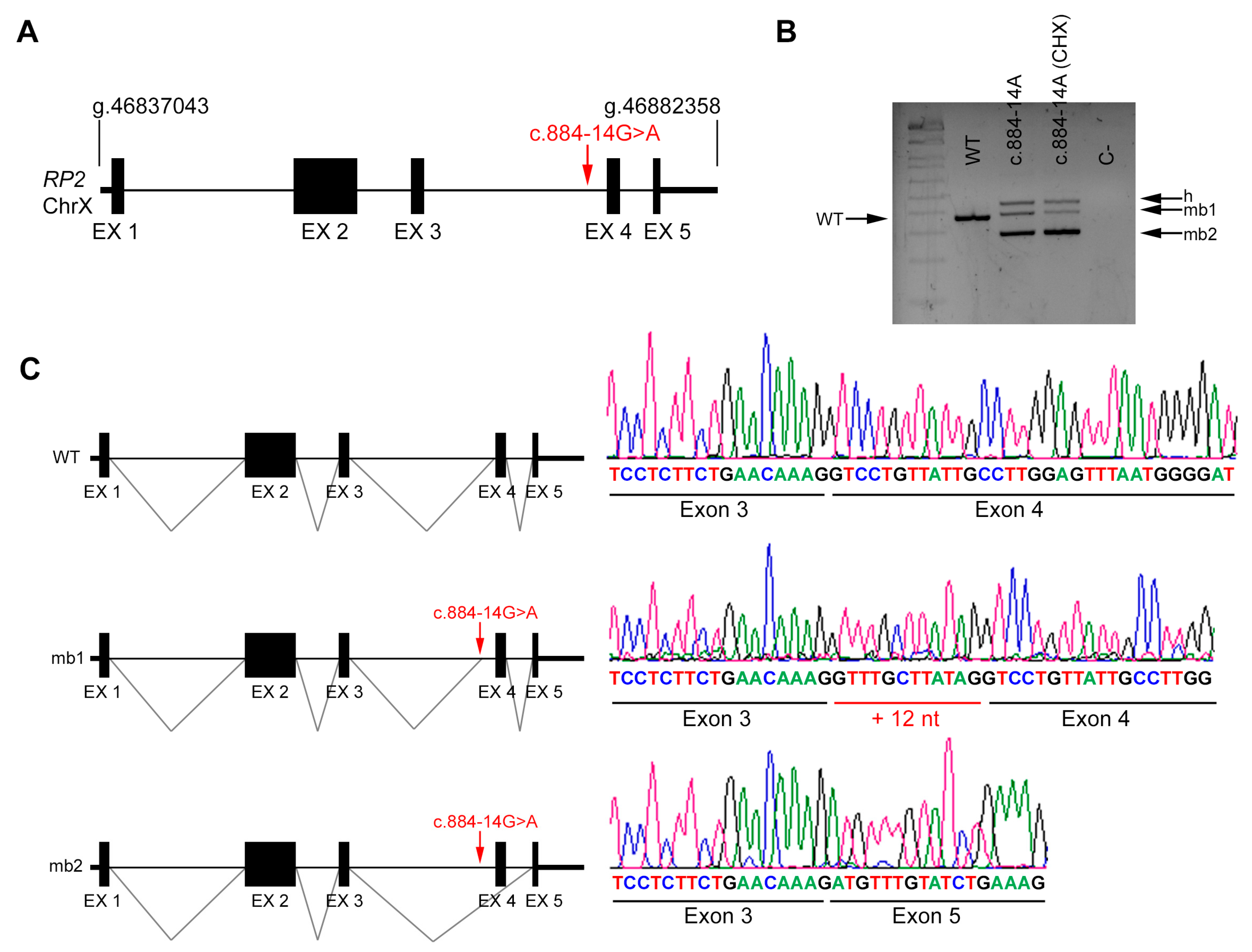
| DBG3 | Index | RP2 (X chrom.) | IVS3 | c.884-14G>A | NA | |
| Unaffected mother | c.884-14G>A | Reference Sequence (WT) | ||||
| DBG4 | Index | POC1B | IVS2 | c.101-3T>G | IVS11 | c.1332+5G>A |
| Unaffected father | c.101-3T>G | Reference Sequence (WT) | ||||
| Unaffected mother | Reference Sequence (WT) | c.1332+5G>A | ||||
| Affected brother | c.101-3T>G | c.1332+5G>A | ||||
| Subject | Gene | Nucleotide Variant | Intron | MAF | HSF | MaxEntScan | NetGene | NNSplice | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | MUT | WT | MUT | WT | MUT | WT | MUT | ||||||||
| DBG1 | ABCA4 | c.4774-9 G>A | IVS33 | ND | AS 79.18 | AS 79.0 | NewAS 79.05 | AS 8.06 | AS 3.04 | NewAS 6.08 | AS 0.15 | AS 0.15 | AS 0.77 | AS NR | NewAS 0.93 |
| DBG2 | ABCA4 | c.4849-8C>G | IVS34 | ND | AS 86.55 | AS 84.0 | NewAS 89.73 | AS 10.9 | AS 4.46 | NewAS 3.64 | AS NR | AS NR | AS 0.98 | AS 0.91 | |
| DBG3 | RP2 | c.884-14G>A | IVS3 | ND | AS 85.41 | AS NR | NewAS 78.72 | AS 8.69 | AS 5.5 | AS 0.90 | AS 0.87 | AS 0.95 | AS 0.92 | NewAS 0.66 | |
| DBG4 | POC1B | c.101-3T>G | IVS2 | 0.0004 | AS 79.56 | AS 76.9 | NewAS 79.84 | AS 7.64 | AS −1.4 | AS 0.15 | AS NR | AS 0.94 | AS NR | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toulis, V.; Cortés-González, V.; de Castro-Miró, M.; Ferraz Sallum, J.; Català-Mora, J.; Villanueva-Mendoza, C.; Ciccioli, M.; Gonzàlez-Duarte, R.; Valero, R.; Marfany, G. Increasing the Genetic Diagnosis Yield in Inherited Retinal Dystrophies: Assigning Pathogenicity to Novel Non-canonical Splice Site Variants. Genes 2020, 11, 378. https://doi.org/10.3390/genes11040378
Toulis V, Cortés-González V, de Castro-Miró M, Ferraz Sallum J, Català-Mora J, Villanueva-Mendoza C, Ciccioli M, Gonzàlez-Duarte R, Valero R, Marfany G. Increasing the Genetic Diagnosis Yield in Inherited Retinal Dystrophies: Assigning Pathogenicity to Novel Non-canonical Splice Site Variants. Genes. 2020; 11(4):378. https://doi.org/10.3390/genes11040378
Chicago/Turabian StyleToulis, Vasileios, Vianney Cortés-González, Marta de Castro-Miró, Juliana Ferraz Sallum, Jaume Català-Mora, Cristina Villanueva-Mendoza, Marcela Ciccioli, Roser Gonzàlez-Duarte, Rebeca Valero, and Gemma Marfany. 2020. "Increasing the Genetic Diagnosis Yield in Inherited Retinal Dystrophies: Assigning Pathogenicity to Novel Non-canonical Splice Site Variants" Genes 11, no. 4: 378. https://doi.org/10.3390/genes11040378
APA StyleToulis, V., Cortés-González, V., de Castro-Miró, M., Ferraz Sallum, J., Català-Mora, J., Villanueva-Mendoza, C., Ciccioli, M., Gonzàlez-Duarte, R., Valero, R., & Marfany, G. (2020). Increasing the Genetic Diagnosis Yield in Inherited Retinal Dystrophies: Assigning Pathogenicity to Novel Non-canonical Splice Site Variants. Genes, 11(4), 378. https://doi.org/10.3390/genes11040378