Compound Phenotype Due to Recessive Variants in LARP7 and OTOG Genes Disclosed by an Integrated Approach of SNP-Array and Whole Exome Sequencing


 ,
, 
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genomic DNA Extraction and Quantification
2.2. SNP Array Analysis
2.3. Whole-Exome Sequencing
2.4. Variant Designation
3. Results
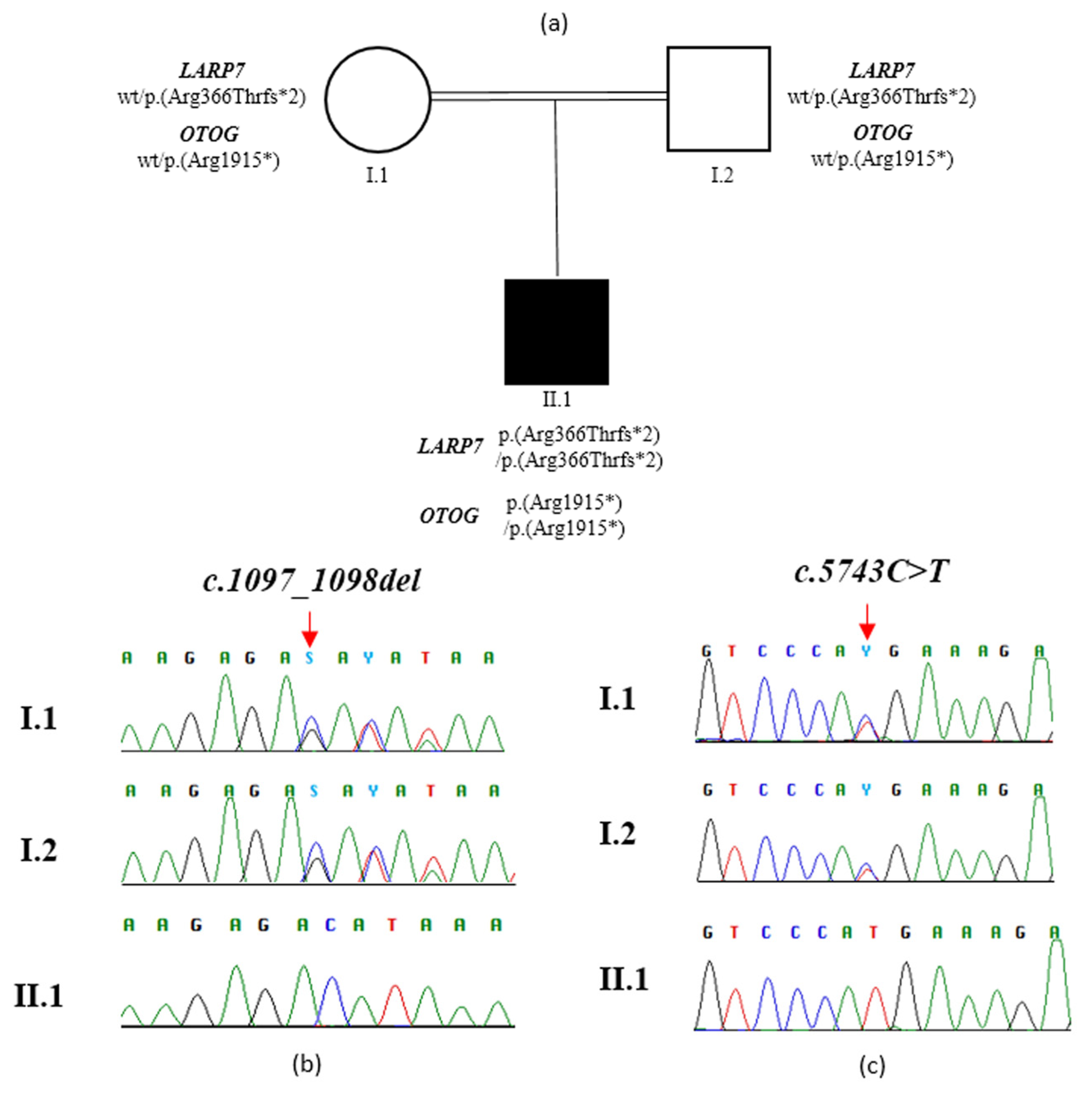
3.1. Clinical Description
3.2. Molecular Findings
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vrijenhoek, T.; Middelburg, E.M.; Monroe, G.R.; van Gassen, K.L.I.; Geenen, J.W.; Hövels, A.M.; Knoers, N.V.; van Amstel, H.K.P.; Frederix, G.W.J. Whole-exome sequencing in intellectual disability; cost before and after a diagnosis. Eur. J. Hum. Genet. 2018, 26, 1566–1571. [Google Scholar] [CrossRef] [Green Version]
- Perucca, P.; Scheffer, I.E.; Harvey, A.S.; James, P.A.; Lunke, S.; Thorne, N.; Gaff, C.; Regan, B.M.; Damiano, J.A.; Hildebrand, M.S.; et al. Real-world utility of whole exome sequencing with targeted gene analysis for focal epilepsy. Epilepsy Res. 2017, 131, 1–8. [Google Scholar] [CrossRef]
- Lenfant, C.; Baz, P.; Degavre, A.; Philippi, A.; Senée, V.; Vandiedonck, C.; Derbois, C.; Nicolino, M.; Zalloua, P.; Julier, C. Juvenile-Onset Diabetes and Congenital Cataract: “Double-Gene” Mutations Mimicking a Syndromic Diabetes Presentation. Genes 2017, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Alazami, A.M.; Al-Owain, M.; Alzahrani, F.; Shuaib, T.; Al-Shamrani, H.; Al-Falki, Y.H.; Al-Qahtani, S.M.; Alsheddi, T.; Colak, D.; Alkuraya, F.S. Loss of function mutation in LARP7, chaperone of 7SK ncRNA, causes a syndrome of facial dysmorphism, intellectual disability, and primordial dwarfism. Hum. Mut. 2012, 33, 1429–1434. [Google Scholar] [CrossRef]
- Ivanovski, I.; Caraffi, S.G.; Magnani, E.; Rosato, S.; Pollazzon, M.; Matalonga, L.; Piana, S.; Nicoli, D.; Baldo, C.; Bernasconi, S.; et al. Alazami syndrome: The first case of papillary thyroid carcinoma. J. Hum. Genet. 2020, 65, 133–141. [Google Scholar] [CrossRef]
- Simmler, M.C.; Cohen-Salmon, M.; El-Amraoui, A.; Guillaud, L.; Benichou, J.C.; Petit, C.; Panthier, J.J. Targeted disruption of otog results in deafness and severe imbalance. Nat. Genet. 2000, 24, 139–143. [Google Scholar] [CrossRef]
- Schraders, M.; Ruiz-Palmero, L.; Kalay, E.; Oostrik, J.; del Castillo, F.J.; Sezgin, O.; Beynon, A.J.; Strom, T.M.; Pennings, R.J.; Zazo Seco, C.; et al. Mutations of the gene encoding otogelin are a cause of autosomal-recessive nonsyndromic moderate hearing impairment. Am. J. Hum. Genet. 2012, 91, 883–889. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Choi, H.J.; Lee, J.S.; Lee, H.J.; Rim, J.H.; Choi, J.Y.; Gee, H.Y.; Jung, J. A novel early truncation mutation in OTOG causes prelingual mild hearing loss without vestibular dysfunction. Eur. J. Med. Genet. 2019, 62, 81–84. [Google Scholar] [CrossRef]
- Palumbo, O.; Fichera, M.; Palumbo, P.; Rizzo, R.; Mazzolla, E.; Cocuzza, D.M.; Carella, M.; Mattina, T. TBR1 is the candidate gene for intellectual disability in patients with a 2q24.2 interstitial deletion. Am. J. Med. Genet. 2014, 163, 828–833. [Google Scholar] [CrossRef]
- Kearney, H.M.; Thorland, E.C.; Brown, K.K.; Quintero-Rivera, F.; South, S.T.; Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet. Med. 2011, 13, 680–685. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Glusman, G.; Caballero, J.; Mauldin, D.E.; Hood, L.; Roach, J.C. KAVIAR: An accessible system for testing SNV novelty. Bioinformatics 2011, 27, 3216–3217. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Non-synonymous and Splice Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Hollink, I.H.; Alfadhel, M.; Al-Wakeel, A.S.; Ababneh, F.; Pfundt, R.; de Man, S.A.; Jamra, R.A.; Rolfs, A.; Bertoli-Avella, A.M.; van de Laar, I.M. Broadening the phenotypic spectrum of pathogenic LARP7 variants: Two cases with intellectual disability, variable growth retardation and distinct facial features. J. Hum. Genet. 2016, 61, 229–233. [Google Scholar] [CrossRef]
- Uchikawa, E.; Natchiar, K.S.; Han, X.; Proux, F.; Roblin, P.; Zhang, E.; Durand, A.; Klaholz, B.P.; Dock-Bregeon, A.C. Structural insight into the mechanism of stabilization of the 7SK small nuclear RNA by LARP7. Nucleic Acids Res. 2015, 43, 3373–3388. [Google Scholar] [CrossRef]
- Eichhorn, C.D.; Chug, R.; Feigon, J. hLARP7 C-terminal domain contains an xRRM that binds the 3’ hairpin of 7SK RNA. Nucleic Acids Res. 2016, 44, 9977–9989. [Google Scholar] [CrossRef] [Green Version]
- Najmabadi, H.; Hu, H.; Garshasbi, M.; Zemojtel, T.; Abedini, S.S.; Chen, W.; Hosseini, M.; Behjati, F.; Haas, S.; Jamali, P.; et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011, 44, 57–63. [Google Scholar] [CrossRef]
- Ling, T.T.; Sorrentino, S. Compound heterozygous variants in the LARP7 gene as a cause of Alazami syndrome in a Caucasian female with significant failure to thrive, short stature, and developmental disability. Am. J. Med. Genet. 2016, 170, 217–219. [Google Scholar] [CrossRef]
- Dateki, S.; Kitajima, T.; Kihara, T.; Watanabe, S.; Yoshiura, K.I.; Moriuchi, H. Novel compound heterozygous variants in the LARP7 gene in a patient with Alazami syndrome. Hum Genome Var. 2018, 5, 18014. [Google Scholar] [CrossRef]
- Imbert-Bouteille, M.; Mau Them, F.T.; Thevenon, J.; Guignard, T.; Gatinois, V.; Riviere, J.B.; Boland, A.; Meyer, V.; Deleuze, J.F.; Sanchez, E.; et al. LARP7 variants and further delineation of the Alazami syndrome phenotypic spectrum among primordial dwarfisms: 2 sisters. Eur. J. Med. Genet. 2019, 62, 161–166. [Google Scholar] [CrossRef]
- Wojcik, M.H.; Linnea, K.; Stoler, J.M.; Rappaport, L. Updating the neurodevelopmental profile of Alazami syndrome: Illustrating the role of developmental assessment in rare genetic disorders. Am. J. Med. Genet. 2019, 179, 1565–1569. [Google Scholar] [CrossRef]
- Goodyear, R.J.; Richardson, G.P. Structure, Function, and Development of the Tectorial Membrane: An Extracellular Matrix Essential for Hearing. Curr. Top. Dev. Biol. 2018, 130, 217–244. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regions of Homozigosity (ISCN 2016) | Size (Mb) | Number of Genes |
|---|---|---|
| arr[GRCh37] 2p25.2p25.1(5573938_10537316)x2 hmz | 4.9 | 40 |
| arr[GRCh37] 2p21p11.2(47453937_89129064)x2 hmz | 41.6 | 317 |
| arr[GRCh37] 2q11.1q22.1(95341387_137166151)x2 hmz | 41.8 | 340 |
| arr[GRCh37] 3p13p11.1(71199337_90485635)x2 hmz | 19.2 | 49 |
| arr[GRCh37] 3q11.1q13.33(93536053_120421935)x2 hmz | 26.8 | 173 |
| arr[GRCh37] 4p15.2p11(21674784_49089181)x2 hmz | 27.4 | 117 |
| arr[GRCh37] 4q11q31.1(52686799_141024195)x2 hmz | 88.3 | 468 |
| arr[GRCh37] 7p14.2p12.2(36814859_50418506)x2 hmz | 13.6 | 98 |
| arr[GRCh37] 7p12.1p11.1(51736013_58019983)x2 hmz | 6.2 | 38 |
| arr[GRCh37] 7q11.21q11.22(62461703_68908285)x2 hmz | 6.4 | 50 |
| arr[GRCh37] 8p23.3p23.1(168483_7011075)x2 hmz | 6.8 | 46 |
| arr[GRCh37] 11p15.5p13(1751363_33420180)x2 hmz | 31.6 | 367 |
| arr[GRCh37] 16p13.3p13.2(6643315_8047081)x2 hmz | 1.4 | 1 |
| arr[GRCh37] 18q12.2q21.1(34083335_43959703)x2 hmz | 9.8 | 33 |
| Chromosome | Start | End | Reference Allele | Alternative Allele | Genotype | Gene | Nucleotide Change | Amino Acid Change | Gene Impact | dbSNP ID | gnomAD_exome Allele Count | TOPMED Allele Count | ExAC_ALL Allele Count |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 | 113568939 | 113568941 | AAG | A | homo_alt | LARP7 NM_015454.2 | c.1097_1098del | p.(Arg366Thrfs*2) | frameshift substitution | rs566464249 | 4/237240 MAF 0.00002 | 6/125568 MAF 0.00005 | 3/113350 0.00003 |
| 11 | 17632554 | 17632554 | C | T | homo_alt | OTOG NM_001277269.1 | c.5743C>T | p.(Arg1915*) | stopgain | rs761287044 | 5/146398 MAF 0.00003 | 2/125568 MAF 0.00002 | 1/15676 0.0001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palumbo, P.; Palumbo, O.; Leone, M.P.; di Muro, E.; Castellana, S.; Bisceglia, L.; Mazza, T.; Carella, M.; Castori, M. Compound Phenotype Due to Recessive Variants in LARP7 and OTOG Genes Disclosed by an Integrated Approach of SNP-Array and Whole Exome Sequencing. Genes 2020, 11, 379. https://doi.org/10.3390/genes11040379
Palumbo P, Palumbo O, Leone MP, di Muro E, Castellana S, Bisceglia L, Mazza T, Carella M, Castori M. Compound Phenotype Due to Recessive Variants in LARP7 and OTOG Genes Disclosed by an Integrated Approach of SNP-Array and Whole Exome Sequencing. Genes. 2020; 11(4):379. https://doi.org/10.3390/genes11040379
Chicago/Turabian StylePalumbo, Pietro, Orazio Palumbo, Maria Pia Leone, Ester di Muro, Stefano Castellana, Luigi Bisceglia, Tommaso Mazza, Massimo Carella, and Marco Castori. 2020. "Compound Phenotype Due to Recessive Variants in LARP7 and OTOG Genes Disclosed by an Integrated Approach of SNP-Array and Whole Exome Sequencing" Genes 11, no. 4: 379. https://doi.org/10.3390/genes11040379
APA StylePalumbo, P., Palumbo, O., Leone, M. P., di Muro, E., Castellana, S., Bisceglia, L., Mazza, T., Carella, M., & Castori, M. (2020). Compound Phenotype Due to Recessive Variants in LARP7 and OTOG Genes Disclosed by an Integrated Approach of SNP-Array and Whole Exome Sequencing. Genes, 11(4), 379. https://doi.org/10.3390/genes11040379