Evolution of Predicted Acid Resistance Mechanisms in the Extremely Acidophilic Leptospirillum Genus

 , and
, and 
Abstract
:1. Introduction
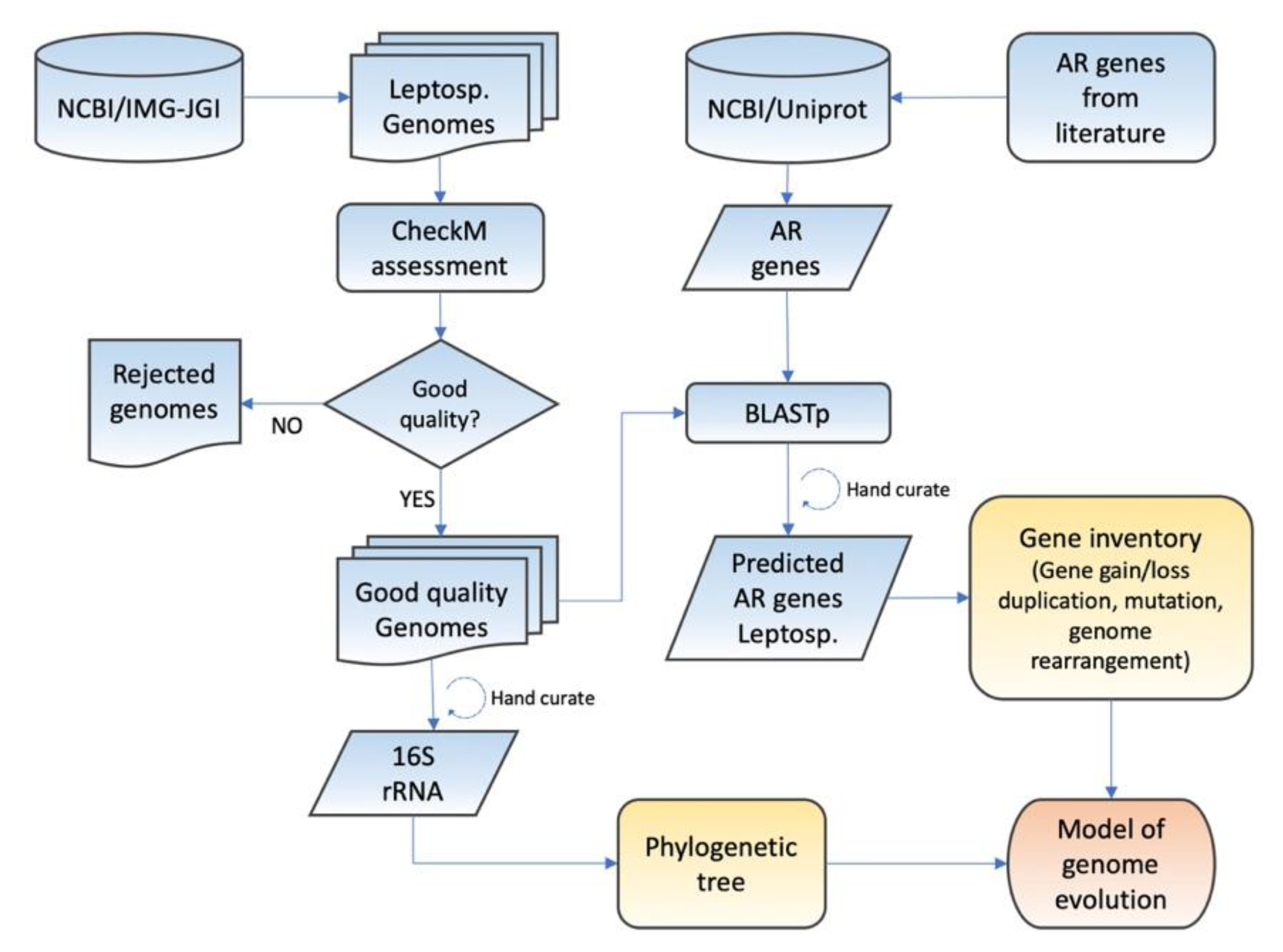
2. Methods
2.1. Genomes and Quality Assessment
2.2. Phylogenetic Analysis
2.3. Prediction of Mobile Genetic Elements and Genome Islands
2.4. Identification of Genes Related to Low pH Resistance
2.5. Evolutionary Pressures on Acid Resistance Genes
2.6. Mapping Evolutionary Events
3. Results and Discussion
3.1. Genomic Features of Leptospirillum Genomes
3.2. Phylogenetic Relatedness between Leptospirillum Species and Other Members of the Nitrospirae Phylum
3.3. Gene Inventories
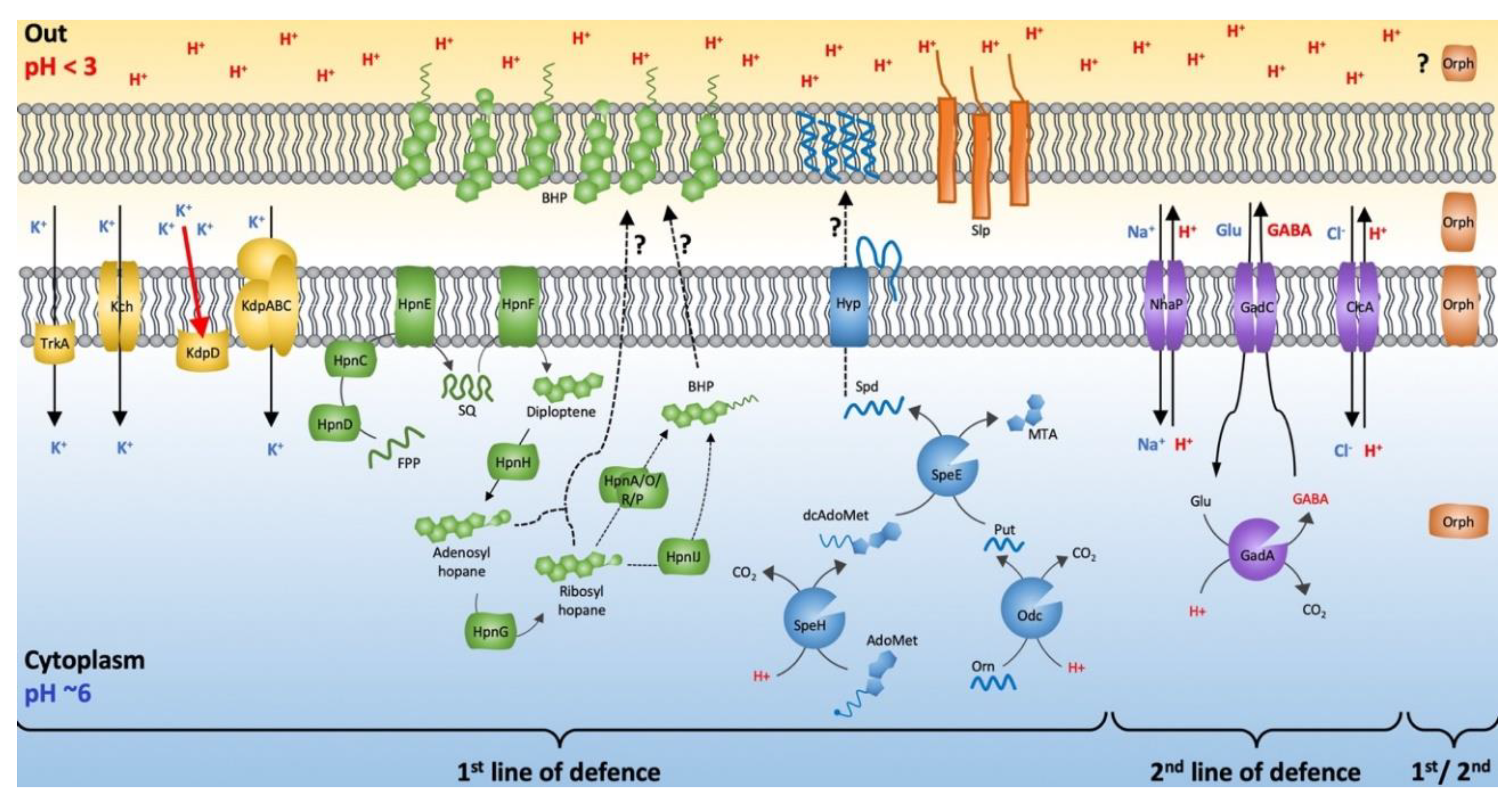
3.4. First Line of Defense
3.4.1. Membrane Potential and Potassium Transporters
3.4.2. Spermidine Biosynthesis and Associated Genes
3.4.3. Hopanoid Biosynthesis
3.4.4. Slp Starvation Lipoprotein
3.5. Second Line of Defense
3.5.1. Proton Antiporters
3.5.2. Gad Decarboxylase
3.6. Model of Leptospirillum Acid Resistance
3.7. Phylogenetic Distribution of Acid Resistance Genes and Their Inferred Evolutionary Trajectories
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Slonczewski, J.L.; Fujisawa, M.; Dopson, M.; Krulwich, T.A. Cytoplasmic pH measurement and homeostasis in bacteria and archaea. Adv. Microb. Physiol. 2009, 55, 1–79. [Google Scholar]
- Tucker, D.L.; Tucker, N.; Conway, T. Gene expression profiling of the pH response in Escherichia coli. J. Bacteriol. 2002, 184, 6551–6558. [Google Scholar] [CrossRef] [Green Version]
- Kanjee, U.; Houry, W.A. Mechanisms of acid resistance in Escherichia coli. Annu. Rev. Microbiol. 2013, 67, 65–81. [Google Scholar] [CrossRef] [Green Version]
- Krulwich, T.A.; Sachs, G.; Padan, E. Molecular aspects of bacterial pH sensing and homeostasis. Nat. Rev. Microbiol. 2011, 9, 330–343. [Google Scholar] [CrossRef] [Green Version]
- Foster, J.W. Escherichia coli acid resistance: Tales of an amateur acidophile. Nat. Rev. Microbiol. 2004, 2, 898–907. [Google Scholar] [CrossRef]
- Zhao, B.; Houry, W.A. Acid stress response in enteropathogenic Gammaproteobacteria: An aptitude for survival. Biochem. Cell Biol. 2010, 88, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Quatrini, R.; Johnson, D.B. (Eds.) Acidophiles: Life in Extremely Acidic Environments; Caister Academic Press: Norfolk, UK, 2016. [Google Scholar] [CrossRef]
- Baker-Austin, C.; Dopson, M. Life in acid: pH homeostasis in acidophiles. Trends Microbiol. 2007, 15, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Hedrich, S.; Schippers, A. Distribution of acidophilic microorganisms in natural and man-made acidic enviroments. In Acidophiles: Life in Extremely Acidic Environments; Quatrini, R., Johnson, D.B., Eds.; Caister Academic Press: Norfolk, UK, 2016; pp. 153–175. [Google Scholar]
- Quatrini, R.; Johnson, D.B. Microbiomes in extremely acidic environments: Functionalities and interactions that allow survival and growth of prokaryotes at low pH. Curr. Opin. Microbiol. 2018, 43, 139–147. [Google Scholar] [CrossRef]
- Zammit, C.M.; Watkin, E.L.J. Adaptation to extreme acidity and osmotic stress. In Acidophiles: Life in Extremely Acidic Environments; Quatrini, R., Johnson, D.B., Eds.; Caister Academic Press: Norfolk, UK, 2016; pp. 49–62. [Google Scholar]
- Buetti-Dinh, A.; Dethlefsen, O.; Friedman, R.; Dopson, M. Transcriptomic analysis reveals how a lack of potassium ions increases Sulfolobus acidocaldarius sensitivity to pH changes. Microbiology (Read. Engl.) 2016, 162, 1422–1434. [Google Scholar] [CrossRef] [PubMed]
- Christel, S.; Herold, M.; Bellenberg, S.; El Hajjami, M.; Buetti-Dinh, A.; Pivkin, I.V.; Sand, W.; Wilmes, P.; Poetsch, A.; Dopson, M. Multi-omics reveals the lifestyle of the acidophilic, mineral-oxidizing model species Leptospirillum ferriphilumT. Appl. Environ. Microbiol. 2018, 84, e02091-17. [Google Scholar] [CrossRef] [Green Version]
- Cholo, M.C.; van Rensburg, E.J.; Osman, A.G.; Anderson, R. Expression of the genes encoding the Trk and Kdp potassium transport systems of Mycobacterium tuberculosis during growth in vitro. Biomed. Res. Int. 2015, 2015, 608682. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.S.; Pedersen, B.P.; Stokes, D.L. Crystal structure of the potassium-importing KdpFABC membrane complex. Nature 2017, 546, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Epstein, W. The roles and regulation of potassium in bacteria. Prog. Nucleic Acid Res. Mol. Biol. 2003, 75, 293–320. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, G. Bioenergetics of the archaebacterium Sulfolobus. Biochim. Et Biophys. Acta 1996, 1277, 163–200. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, I.; Lee, D.; Mackay, B.; Harahuc, L.; Oh, J.K. Effect of various ions, pH, and osmotic pressure on oxidation of elemental sulfur by Thiobacillus thiooxidans. Appl. Environ. Microbiol. 1999, 65, 5163–5168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goulbourne, E.J.; Matin, M.; Zychlinsky, E. Mechanism of delta pH maintenance in active and inactive cells of an obligately acidophilic bacterium. J. Bacteriol. 1986, 166, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Rhee, H.J.; Kim, E.J.; Lee, J.K. Physiological polyamines: Simple primordial stress molecules. J. Cell. Mol. Med. 2007, 11, 685–703. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.H.; Casavant, C.; Hawley, Q.; Addwebi, T.; Call, D.R.; Guard, J. Salmonella Enteritidis strains from poultry exhibit differential responses to acid stress, oxidative stress, and survival in the egg albumen. Foodborne Pathog. Dis. 2012, 9, 258–264. [Google Scholar] [CrossRef] [Green Version]
- Watson, N.; Dunyak, D.S.; Rosey, E.L.; Slonczewski, J.L.; Olson, E.R. Identification of elements involved in transcriptional regulation of the Escherichia coli cad operon by external pH. J. Bacteriol. 1992, 174, 530–540. [Google Scholar] [CrossRef] [Green Version]
- Dela Vega, A.L.; Delcour, A.H. Polyamines decrease Escherichia coli outer membrane permeability. J. Bacteriol. 1996, 178, 3715–3721. [Google Scholar] [CrossRef] [Green Version]
- Hosoya, R.; Yokoyama, Y.; Hamana, K.; Itoh, T. Polyamine analysis within the eubacterial thirteen phyla Acidobacteria, Actinobacteria, Chlorobi, Chloroflexi, Chrysiogenetes, Deferribacteres, Fibrobacteres, Firmicutes, Fusobacteria, Gemmatimonadetes, Nitrospirae, Planctomycetes, and Verrucomicrobia. Microbiol. Cult. Coll. 2006, 22, 21–33. [Google Scholar]
- Li, Q.; Li, N.; Liu, X.; Zhou, Z.; Fang, Y.; Fan, X.; Fu, X.; Liu, Y.D.; Yin, H.Q. Characterization of the acid stress response of Acidithiobacillus ferrooxidans ATCC 23270 based on the method of microarray. J. Biol. Res.-Thessal. 2012, 17, 3–15. [Google Scholar]
- Belin, B.J.; Busset, N.; Giraud, E.; Molinaro, A.; Silipo, A.; Newman, D.K. Hopanoid lipids: From membranes to plant–bacteria interactions. Nat. Rev. Microbiol. 2018, 16, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohlenkamp, C.; Geiger, O. Bacterial membrane lipids: Diversity in structures and pathways. FEMS Microbiol. Rev. 2016, 40, 133–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welander, P.V.; Hunter, R.C.; Zhang, L.; Sessions, A.L.; Summons, R.E.; Newman, D.K. Hopanoids play a role in membrane integrity and pH homeostasis in Rhodopseudomonas palustris TIE-1. J. Bacteriol. 2009, 191, 6145–6156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmerk, C.L.; Welander, P.V.; Hamad, M.A.; Bain, K.L.; Bernards, M.A.; Summons, R.E.; Valvano, M.A. Elucidation of the Burkholderia cenocepacia hopanoid biosynthesis pathway uncovers functions for conserved proteins in hopanoid-producing bacteria. Environ. Microbiol. 2015, 17, 735–750. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, G.; Wu, C.H.; Newman, D.K. The general stress response factor EcfG regulates expression of the C-2 hopanoid methylase HpnP in Rhodopseudomonas palustris TIE-1. J. Bacteriol. 2013, 195, 2490–2498. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.J.; Solbiati, J.O.; Ramamoorthy, G.; Hillerich, B.S.; Seidel, R.D.; Cronan, J.E.; Almo, S.C.; Poulter, C.D. Biosynthesis of squalene from farnesyl diphosphate in bacteria: Three steps catalyzed by three enzymes. ACS Cent. Sci. 2015, 1, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.S.; Albrecht, H.L.; Dawson, K.S.; Schaperdoth, I.; Freeman, K.H.; Pi, Y.; Pearson, A.; Macalady, J.L. Community genomic analysis of an extremely acidophilic sulfur-oxidizing biofilm. ISME J. 2012, 6, 158–170. [Google Scholar] [CrossRef]
- Mangold, S.; Rao Jonna, V.; Dopson, M. Response of Acidithiobacillus caldus toward suboptimal pH conditions. Extremophiles 2013, 17, 689–696. [Google Scholar] [CrossRef]
- Chen, L.X.; Hu, M.; Huang, L.N.; Hua, Z.S.; Kuang, J.L.; Li, S.J.; Shu, W.S. Comparative metagenomic and metatranscriptomic analyses of microbial communities in acid mine drainage. ISME J. 2015, 9, 1579–1592. [Google Scholar] [CrossRef] [PubMed]
- Hommais, F.; Krin, E.; Coppee, J.Y.; Lacroix, C.; Yeramian, E.; Danchin, A.; Bertin, P. GadE (YhiE): A novel activator involved in the response to acid environment in Escherichia coli. Microbiology (Read. Engl.) 2004, 150, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merrell, D.S.; Camilli, A. Acid tolerance of gastrointestinal pathogens. Curr. Opin. Microbiol. 2002, 5, 51–55. [Google Scholar] [CrossRef]
- Alexander, D.M.; St John, A.C. Characterization of the carbon starvation-inducible and stationary phase-inducible gene slp encoding an outer membrane lipoprotein in Escherichia coli. Mol. Microbiol. 1994, 11, 1059–1071. [Google Scholar] [CrossRef]
- Mates, A.K.; Sayed, A.K.; Foster, J.W. Products of the Escherichia coli acid fitness island attenuate metabolite stress at extremely low pH and mediate a cell density-dependent acid resistance. J. Bacteriol. 2007, 189, 2759–2768. [Google Scholar] [CrossRef] [Green Version]
- Alexander, B.; Leach, S.; Ingledew, W.J. The relationship between chemiosmotic parameters and sensitivity to anions and organic acids in the acidophile Thiobacillus ferrooxidans. J. Gen. Microbiol. 1987, 133, 1171–1179. [Google Scholar] [CrossRef] [Green Version]
- Padan, E.; Tzubery, T.; Herz, K.; Kozachkov, L.; Rimon, A.; Galili, L. NhaA of Escherichia coli, as a model of a pH-regulated Na+/H+ antiporter. Biochim. Biophys. Acta-Bioenerg. 2004, 1658, 2–13. [Google Scholar] [CrossRef] [Green Version]
- Accardi, A.; Walden, M.; Nguitragool, W.; Jayaram, H.; Williams, C.; Miller, C. Separate ion pathways in a Cl-/H+ exchanger. J. Gen. Physiol. 2005, 126, 563–570. [Google Scholar] [CrossRef] [Green Version]
- Iyer, R.; Iverson, T.M.; Accardi, A.; Miller, C. A biological role for prokaryotic ClC chloride channels. Nature 2002, 419, 715–718. [Google Scholar] [CrossRef]
- Richard, H.; Foster, J.W. Escherichia coli glutamate- and arginine-dependent acid resistance systems increase internal pH and reverse transmembrane potential. J. Bacteriol. 2004, 186, 6032–6041. [Google Scholar] [CrossRef] [Green Version]
- McLaggan, D.; Keyhan, M.; Matin, A. Chloride transport pathways and their bioenergetic implications in the obligate acidophile Bacillus coagulans. J. Bacteriol. 1990, 172, 1485–1490. [Google Scholar] [CrossRef] [Green Version]
- Feehily, C.; Karatzas, K.A. Role of glutamate metabolism in bacterial responses towards acid and other stresses. J. Appl. Microbiol. 2013, 114, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Mi, S.; Song, J.; Lin, J.; Che, Y.; Zheng, H.; Lin, J. Complete genome of Leptospirillum ferriphilum ML-04 provides insight into its physiology and environmental adaptation. J. Microbiol. 2011, 49, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Coram, N.J.; Rawlings, D.E. Molecular relationship between two groups of the genus Leptospirillum and the finding that Leptospirillum ferriphilum sp. nov. dominates South African commercial biooxidation tanks that operate at 40°C. Appl. Environ. Microbiol. 2002, 68, 838–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hippe, H. Leptospirillium gen. nov (ex Markoysan 1972), nom. rev., including Leptospirillium ferrooxidans sp nov (ex Markoysan 1972), nom. rev. and Leptospirillium thermoferrooxidans sp nov (Golovacheva et al. 1992). Int. J. Syst. Evol. Microbiol. 2000, 50, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Bond, P.L.; Smriga, S.P.; Banfield, J.F. Phylogeny of microorganisms populating a thick, subaerial, predominantly lithotrophic biofilm at an extreme acid mine drainage site. Appl. Environ. Microbiol. 2000, 66, 3842–3849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ram, R.J.; VerBerkmoes, N.C.; Thelen, M.P.; Tyson, G.W.; Baker, B.J.; Blake, R.C.; Shah, M.; Hettich, R.L.; Banfield, J.F. Community proteomics of a natural microbial biofilm. Science (N. Y. NY) 2005, 308, 1915–1920. [Google Scholar] [CrossRef]
- Tyson, G.W.; Lo, I.; Baker, B.J.; Allen, E.E.; Hugenholtz, P.; Banfield, J.F. Genome-directed isolation of the key nitrogen fixer Leptospirillum ferrodiazotrophum sp. nov. from an acidophilic microbial community. Appl. Environ. Microbiol. 2005, 71, 6319–6324. [Google Scholar] [CrossRef] [Green Version]
- Goltsman, D.S.; Denef, V.J.; Singer, S.W.; VerBerkmoes, N.C.; Lefsrud, M.; Mueller, R.S.; Dick, G.J.; Sun, C.L.; Wheeler, K.E.; Zemla, A.; et al. Community genomic and proteomic analyses of chemoautotrophic iron-oxidizing “Leptospirillum rubarum” (Group II) and “Leptospirillum ferrodiazotrophum” (Group III) bacteria in acid mine drainage biofilms. Appl. Environ. Microbiol. 2009, 75, 4599–4615. [Google Scholar] [CrossRef] [Green Version]
- Wilmes, P.; Remis, J.P.; Hwang, M.; Auer, M.; Thelen, M.P.; Banfield, J.F. Natural acidophilic biofilm communities reflect distinct organismal and functional organization. ISME J. 2009, 3, 266–270. [Google Scholar] [CrossRef]
- Belnap, C.P.; Pan, C.; VerBerkmoes, N.C.; Power, M.E.; Samatova, N.F.; Carver, R.L.; Hettich, R.L.; Banfield, J.F. Cultivation and quantitative proteomic analyses of acidophilic microbial communities. ISME J. 2010, 4, 520–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belnap, C.P.; Pan, C.; Denef, V.J.; Samatova, N.F.; Hettich, R.L.; Banfield, J.F. Quantitative proteomic analyses of the response of acidophilic microbial communities to different pH conditions. ISME J. 2011, 5, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Saglam, E.S.; Akcay, M.; Colak, D.N.; Inan Bektas, K.; Belduz, A.O. Generation of acid mine drainage around the Karaerik copper mine (Espiye, Giresun, NE Turkey): Implications from the bacterial population in the Acisu effluent. Extremophiles 2016, 20, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Schrenk, M.O.; Edwards, K.J.; Goodman, R.M.; Hamers, R.J.; Banfield, J.F. Distribution of Thiobacillus ferrooxidans and Leptospirillium ferrooxidans: Implications for generation of acid mine drainage. Science (N. Y. NY) 1998, 279, 1519–1522. [Google Scholar] [CrossRef] [Green Version]
- Baker, B.J.; Banfield, J.F. Microbial communities in acid mine drainage. FEMS Microbiol. Ecol. 2003, 44, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Issotta, F.; Galleguillos, P.A.; Moya-Beltrán, A.; Davis-Belmar, C.S.; Rautenbach, G.; Covarrubias, P.C.; Acosta, M.; Ossandon, F.J.; Contador, Y.; Holmes, D.S.; et al. Draft genome sequence of chloride-tolerant Leptospirillum ferriphilum Sp-Cl from industrial bioleaching operations in northern Chile. Stand. Genom. Sci. 2016, 11, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.G.; Peplies, J.; Glockner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [Green Version]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef] [Green Version]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Turenne, C.Y.; Tschetter, L.; Wolfe, J.; Kabani, A. Necessity of quality-controlled 16S rRNA gene sequence databases: Identifying nontuberculous Mycobacterium species. J. Clin. Microbiol. 2001, 39, 3637–3648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.i.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Tommaso, P.; Moretti, S.; Xenarios, I.; Orobitg, M.; Montanyola, A.; Chang, J.M.; Taly, J.F.; Notredame, C. T-Coffee: A web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 2011, 39, W13–W17. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Riadi, G.; Medina-Moenne, C.; Holmes, D.S. TnpPred: A web service for the robust prediction of prokaryotic transposases. Comp. Funct. Genom. 2012, 2012, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef] [Green Version]
- Varani, A.M.; Siguier, P.; Gourbeyre, E.; Charneau, V.; Chandler, M. ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol 2011, 12, R30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waack, S.; Keller, O.; Asper, R.; Brodag, T.; Damm, C.; Fricke, W.F.; Surovcik, K.; Meinicke, P.; Merkl, R. Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinform. 2006, 7, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [Green Version]
- Carver, T.; Harris, S.R.; Berriman, M.; Parkhill, J.; McQuillan, J.A. Artemis: An integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics (Oxf. Engl.) 2012, 28, 464–469. [Google Scholar] [CrossRef] [Green Version]
- Casiano-Colon, A.; Marquis, R.E. Role of the arginine deiminase system in protecting oral bacteria and an enzymatic basis for acid tolerance. Appl. Environ. Microbiol. 1988, 54, 1318–1324. [Google Scholar] [CrossRef] [Green Version]
- Gajiwala, K.S.; Burley, S.K. HDEA, a periplasmic protein that supports acid resistance in pathogenic enteric bacteria. J. Mol. Biol. 2000, 295, 605–612. [Google Scholar] [CrossRef]
- Jeong, K.C.; Hung, K.F.; Baumler, D.J.; Byrd, J.J.; Kaspar, C.W. Acid stress damage of DNA is prevented by Dps binding in Escherichia coli O157:H7. BMC Microbiol. 2008, 8, 181. [Google Scholar] [CrossRef] [Green Version]
- Leverrier, P.; Vissers, J.P.; Rouault, A.; Boyaval, P.; Jan, G. Mass spectrometry proteomic analysis of stress adaptation reveals both common and distinct response pathways in Propionibacterium freudenreichii. Arch. Microbiol. 2004, 181, 215–230. [Google Scholar] [CrossRef]
- Lund, P.; Tramonti, A.; De Biase, D. Coping with low pH: Molecular strategies in neutralophilic bacteria. FEMS Microbiol. Rev. 2014, 38, 1091–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mujacic, M.; Baneyx, F. Regulation of Escherichia coli hchA, a stress-inducible gene encoding molecular chaperone Hsp31. Mol. Microbiol. 2006, 60, 1576–1589. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.; Ladero, V.; Alvarez, M.A.; Lucas, P.M. Putrescine production via the ornithine decarboxylation pathway improves the acid stress survival of Lactobacillus brevis and is part of a horizontally transferred acid resistance locus. Int. J. Food Microbiol. 2014, 175, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Soksawatmaekhin, W.; Kuraishi, A.; Sakata, K.; Kashiwagi, K.; Igarashi, K. Excretion and uptake of cadaverine by CadB and its physiological functions in Escherichia coli. Mol. Microbiol. 2004, 51, 1401–1412. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, T.D.; Stephens, R.M. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 1990, 18, 6097–6100. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics (Oxf. Engl.) 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Jeffares, D.C.; Tomiczek, B.; Sojo, V.; dos Reis, M. A beginners guide to estimating the non-synonymous to synonymous rate ratio of all protein-coding genes in a genome. Methods Mol. Biol. (Cliftonn. J.) 2015, 1201, 65–90. [Google Scholar] [CrossRef]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charif, D.; Lobry, J. SeqinR 1.0-2: A contributed package to the R Project for statistical computing devoted to biological sequences retrieval and analysis. In Structural Approaches to Sequence Evolution; Bastolla, U., Porto, M., Roman, H.E., Vendruscolo, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 207–232. [Google Scholar]
- Li, W.H.; Wu, C.I.; Luo, C.C. A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol. Biol. Evol. 1985, 2, 150–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, O.; Ashkenazy, H.; Belinky, F.; Huchon, D.; Pupko, T. GLOOME: Gain loss mapping engine. Bioinformatics (Oxf. Engl.) 2010, 26, 2914–2915. [Google Scholar] [CrossRef] [PubMed]
- Cohen, O.; Pupko, T. Inference of gain and loss events from phyletic patterns using stochastic mapping and maximum parsimony--a simulation study. Genome Biol. Evol. 2011, 3, 1265–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimura, R.; Sato, Y.; Nishizawa, T.; Oshima, K.; Kim, S.W.; Hattori, M.; Kamijo, T.; Ohta, H. Complete genome sequence of Leptospirillum ferrooxidans strain C2-3, isolated from a fresh volcanic ash deposit on the island of Miyake, Japan. J. Bacteriol. 2012, 194, 4122–4123. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Liang, Y.; Yin, H.; Xiao, Y.; Guo, X.; Xu, Y.; Hu, Q.; Liu, H.; Liu, X. Effects of arsenite resistance on the growth and functional gene expression of Leptospirillum ferriphilum and Acidithiobacillus thiooxidans in pure culture and coculture. Biomed. Res. Int. 2015, 2015, 203197. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Liu, X.; Liang, Y.; Xiao, Y.; Ma, L.; Guo, X.; Miao, B.; Liu, H.; Peng, D.; Huang, W.; et al. Comparative genomics unravels the functional roles of co-occurring acidophilic bacteria in bioleaching heaps. Front Microbiol. 2017, 8, 790. [Google Scholar] [CrossRef] [Green Version]
- Ferrer, A.; Bunk, B.; Spröer, C.; Biedendieck, R.; Valdés, N.; Jahn, M.; Jahn, D.; Orellana, O.; Levicán, G. Complete genome sequence of the bioleaching bacterium Leptospirillum sp. group II strain CF-1. J. Biotechnol. 2016, 222, 21–22. [Google Scholar] [CrossRef]
- Denef, V.J.; Banfield, J.F. In situ evolutionary rate measurements show ecological success of recently emerged bacterial hybrids. Science (N. Y. NY) 2012, 336, 462–466. [Google Scholar] [CrossRef]
- Simmons, S.L.; Dibartolo, G.; Denef, V.J.; Goltsman, D.S.; Thelen, M.P.; Banfield, J.F. Population genomic analysis of strain variation in Leptospirillum group II bacteria involved in acid mine drainage formation. PLos Biol. 2008, 6, e177. [Google Scholar] [CrossRef]
- Goltsman, D.S.; Dasari, M.; Thomas, B.C.; Shah, M.B.; VerBerkmoes, N.C.; Hettich, R.L.; Banfield, J.F. New group in the Leptospirillum clade: Cultivation-independent community genomics, proteomics, and transcriptomics of the new species “Leptospirillum group IV UBA BS”. Appl. Environ. Microbiol. 2013, 79, 5384–5393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Niu, J.; Liang, Y.; Liu, X.; Yin, H. Metagenome-scale analysis yields insights into the structure and function of microbial communities in a copper bioleaching heap. BMC Genet. 2016, 17, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liljeqvist, M.; Ossandon, F.J.; González, C.; Rajan, S.; Stell, A.; Valdes, J.; Holmes, D.S.; Dopson, M. Metagenomic analysis reveals adaptations to a cold-adapted lifestyle in a low-temperature acid mine drainage stream. FEMS Microb. Ecol. 2015, 91. [Google Scholar] [CrossRef]
- Saier, M.H., Jr.; Reddy, B.L. Holins in bacteria, eukaryotes, and archaea: Multifunctional xenologues with potential biotechnological and biomedical applications. J. Bacteriol. 2015, 197, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Dubnau, D.; Blokesch, M. Mechanisms of DNA uptake by naturally competent bacteria. Annu. Rev. Genet. 2019, 53, 217–237. [Google Scholar] [CrossRef] [PubMed]
- Romling, U.; Gomelsky, M.; Galperin, M.Y. C-di-GMP: The dawning of a novel bacterial signalling system. Mol. Microbiol. 2005, 57, 629–639. [Google Scholar] [CrossRef]
- Diaz, M.; Castro, M.; Copaja, S.; Guiliani, N. Biofilm formation by the acidophile bacterium Acidithiobacillus thiooxidans involves c-di-GMP pathway and Pel exopolysaccharide. Genes 2018, 9, 113. [Google Scholar] [CrossRef] [Green Version]
- Moya-Beltran, A.; Rojas-Villalobos, C.; Diaz, M.; Guiliani, N.; Quatrini, R.; Castro, M. Nucleotide second messenger-based signaling in extreme acidophiles of the Acidithiobacillus species complex: Partition between the core and variable gene complements. Front Microbiol. 2019, 10, 381. [Google Scholar] [CrossRef]
- Jenal, U.; Reinders, A.; Lori, C. Cyclic di-GMP: Second messenger extraordinaire. Nat. Rev. Microbiol. 2017, 15, 271–284. [Google Scholar] [CrossRef] [Green Version]
- Zuckert, W.R. Secretion of bacterial lipoproteins: Through the cytoplasmic membrane, the periplasm and beyond. Biochim. Et Biophys. Acta 2014, 1843, 1509–1516. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Guinn, E.J.; Buechel, E.; Wong, R.; Sengupta, R.; Shkel, I.A.; Record, M.T., Jr. Basis of protein stabilization by K glutamate: Unfavorable interactions with carbon, oxygen groups. Biophys. J. 2016, 111, 1854–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, Z.S.; Han, Y.J.; Chen, L.X.; Liu, J.; Hu, M.; Li, S.J.; Kuang, J.L.; Chain, P.S.; Huang, L.N.; Shu, W.S. Ecological roles of dominant and rare prokaryotes in acid mine drainage revealed by metagenomics and metatranscriptomics. ISME J. 2015, 9, 1280–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosier, A.C.; Li, Z.; Thomas, B.C.; Hettich, R.L.; Pan, C.; Banfield, J.F. Elevated temperature alters proteomic responses of individual organisms within a biofilm community. ISME J. 2015, 9, 180–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullrich, S.R.; González, C.; Poehlein, A.; Tischler, J.S.; Daniel, R.; Schlömann, M.; Holmes, D.S.; Muhling, M. Gene loss and horizontal gene transfer contributed to the genome evolution of the extreme acidophile “Ferrovum”. Front Microbiol. 2016, 7. [Google Scholar] [CrossRef]
- Feng, S.; Yang, H.; Wang, W. System-level understanding of the potential acid-tolerance components of Acidithiobacillus thiooxidans ZJJN-3 under extreme acid stress. Extremophiles 2015, 19, 1029–1039. [Google Scholar] [CrossRef]
- Colman, D.R.; Poudel, S.; Hamilton, T.L.; Havig, J.R.; Selensky, M.J.; Shock, E.L.; Boyd, E.S. Geobiological feedbacks and the evolution of thermoacidophiles. ISME J. 2018, 12, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Fitch, W.M. Toward defining the course of evolution: Minimum change for a specific tree topology. Syst. Zool 1971, 20, 406–416. [Google Scholar] [CrossRef]
- Mirkin, B.G.; Fenner, T.I.; Galperin, M.Y.; Koonin, E.V. Algorithms for computing parsimonious evolutionary scenarios for genome evolution, the last universal common ancestor and dominance of horizontal gene transfer in the evolution of prokaryotes. BMC Evol. Biol. 2003, 3, 2. [Google Scholar] [CrossRef]
- Vialle, R.A.; Tamuri, A.U.; Goldman, N. Alignment modulates ancestral sequence reconstruction accuracy. Mol. Biol. Evol. 2018, 35, 1783–1797. [Google Scholar] [CrossRef] [Green Version]
- Novichkov, P.S.; Omelchenko, M.V.; Gelfand, M.S.; Mironov, A.A.; Wolf, Y.I.; Koonin, E.V. Genome-wide molecular clock and horizontal gene transfer in bacterial evolution. J. Bacteriol. 2004, 186, 6575–6585. [Google Scholar] [CrossRef] [Green Version]
- Guiliani, N.; Jerez, C.A. Molecular cloning, sequencing, and expression of omp-40, the gene coding for the major outer membrane protein from the acidophilic bacterium Thiobacillus ferrooxidans. Appl. Environ. Microbiol. 2000, 66, 2318–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, A.; Valenzuela, L.; Beard, S.; Mackey, A.J.; Shabanowitz, J.; Hunt, D.F.; Jerez, C.A. Periplasmic proteins of the extremophile Acidithiobacillus ferrooxidans: A high throughput proteomics analysis. Mol Cell Proteom. 2007, 6, 2239–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome | Size (Mb) | # Predicted Genes | G + C (%) | pH | Temp (°C) | Status 1 | NCBI Accession 2 | Geographical Origin | Refs |
|---|---|---|---|---|---|---|---|---|---|
| Leptospirillum ferrooxidans C2-3 | 2.5 | 2587 | 50.0 | 1.8 | 30 | C | NC_017094.1 | Mount Oyama, Miyake, Japan | [99] |
| Leptospirillum ferriphilum DSM 14647 | 2.6 | 2687 | 54.1 | 1.4–1.8 | 37 | C | NZ_OBMB00000000.1 | Enrichment culture, Peru | [13] |
| Leptospirillum ferriphilum ML-04 | 2.4 | 2475 | 54.6 | 2.5 | 40 | C | NC_018649.1 | Sulfide hot spring, Yunnan, China | [46] |
| Leptospirillum ferriphilum YSK | 2.3 | 2361 | 54.1 | 1.6 | 40 | C | NZ_CP007243.1 | Dexing copper mine, JiangXi, China | [100] |
| Leptospirillum ferriphilum DX | 2.3 | 2381 | 54.5 | 1.5 | 40 | D | NZ_MPOJ00000000.1 | Dexing copper mine, JiangXi, China | [101] |
| Leptospirillum ferriphilum ZJ | 2.3 | 2449 | 54.7 | 1.5 | 40 | D | NZ_MPOK00000000.1 | Zijinshan copper mine, Fujian, China | [101] |
| Leptospirillum ferriphilum Sp-Cl | 2.4 | 2552 | 54.4 | 1.5 | 37 | D | NZ_LGSH00000000.1 | Spence mine, Chile | [59] |
| Leptospirillum sp. “CF-1” | 2.7 | 2731 | 54.6 | 1.6–1.7 | 40 | C | NZ_CP012147.1 | Iron Mountain, CA, USA | [102] |
| Leptospirillum sp. “C75” | 2.6 | 2528 | 54.4 | 0.7–1.2 | 40–43 | D | GCF_000262365.1 | Iron Mountain, CA, USA | [103] |
| Leptospirillum sp. “5-way CG” | 2.7 | 2633 | 51.5 | 0.8 | 42 | D | DS995259.1–DS995275.1 | Iron Mountain, CA, USA | [104] |
| “Leptospirillum rubarum” | 2.6 | 2654 | 54.7 | 1.1 | 41 | D | GCA_000205145.2 | Iron Mountain, CA, USA | [52] |
| “Leptospirillum ferrodiazotrophum” | 2.8 | 2727 | 57.5 | 1.1 | 41 | D | GG693851.1–GG693892.1 | Iron Mountain, CA, USA | [52] |
| Nitrospira marina Nb-295 | 4.6 | 4276 | 50 | 6.4–7.5 | 30 | D | 2596583682 | Gulf of Maine, USA | ‡ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vergara, E.; Neira, G.; González, C.; Cortez, D.; Dopson, M.; Holmes, D.S. Evolution of Predicted Acid Resistance Mechanisms in the Extremely Acidophilic Leptospirillum Genus. Genes 2020, 11, 389. https://doi.org/10.3390/genes11040389
Vergara E, Neira G, González C, Cortez D, Dopson M, Holmes DS. Evolution of Predicted Acid Resistance Mechanisms in the Extremely Acidophilic Leptospirillum Genus. Genes. 2020; 11(4):389. https://doi.org/10.3390/genes11040389
Chicago/Turabian StyleVergara, Eva, Gonzalo Neira, Carolina González, Diego Cortez, Mark Dopson, and David S. Holmes. 2020. "Evolution of Predicted Acid Resistance Mechanisms in the Extremely Acidophilic Leptospirillum Genus" Genes 11, no. 4: 389. https://doi.org/10.3390/genes11040389
APA StyleVergara, E., Neira, G., González, C., Cortez, D., Dopson, M., & Holmes, D. S. (2020). Evolution of Predicted Acid Resistance Mechanisms in the Extremely Acidophilic Leptospirillum Genus. Genes, 11(4), 389. https://doi.org/10.3390/genes11040389