Genotyping by RAD Sequencing Analysis Assessed the Genetic Distinctiveness of Experimental Lines and Narrowed down the Genomic Region Responsible for Leaf Shape in Endive (Cichorium endivia L.)





Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. SSR-Based Genotyping by Heterologous Chicory-Derived Microsatellites
2.3. SNP-Based Genotyping by RAD-Seq Analysis
2.4. SNP Marker Validation
3. Results
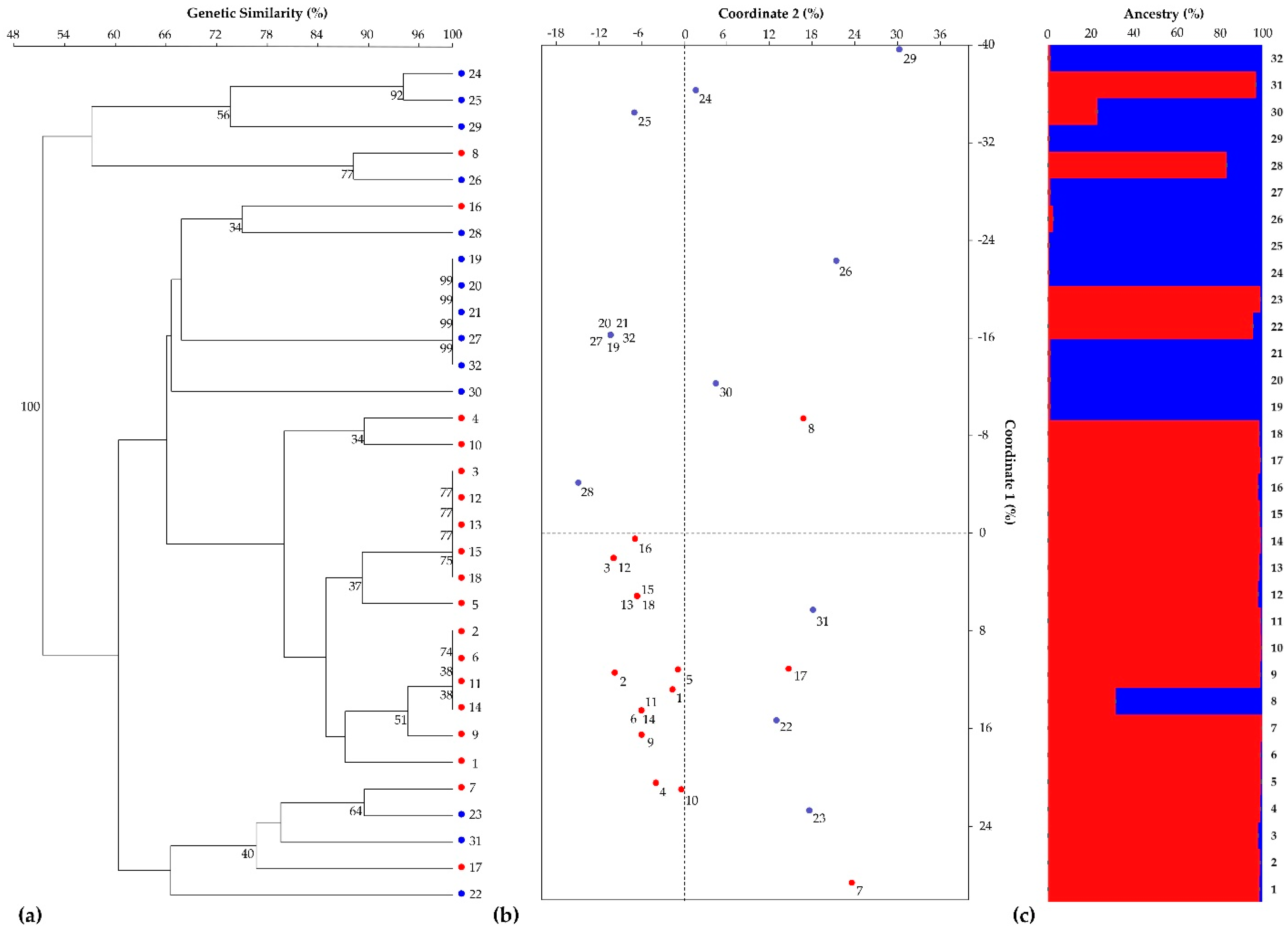
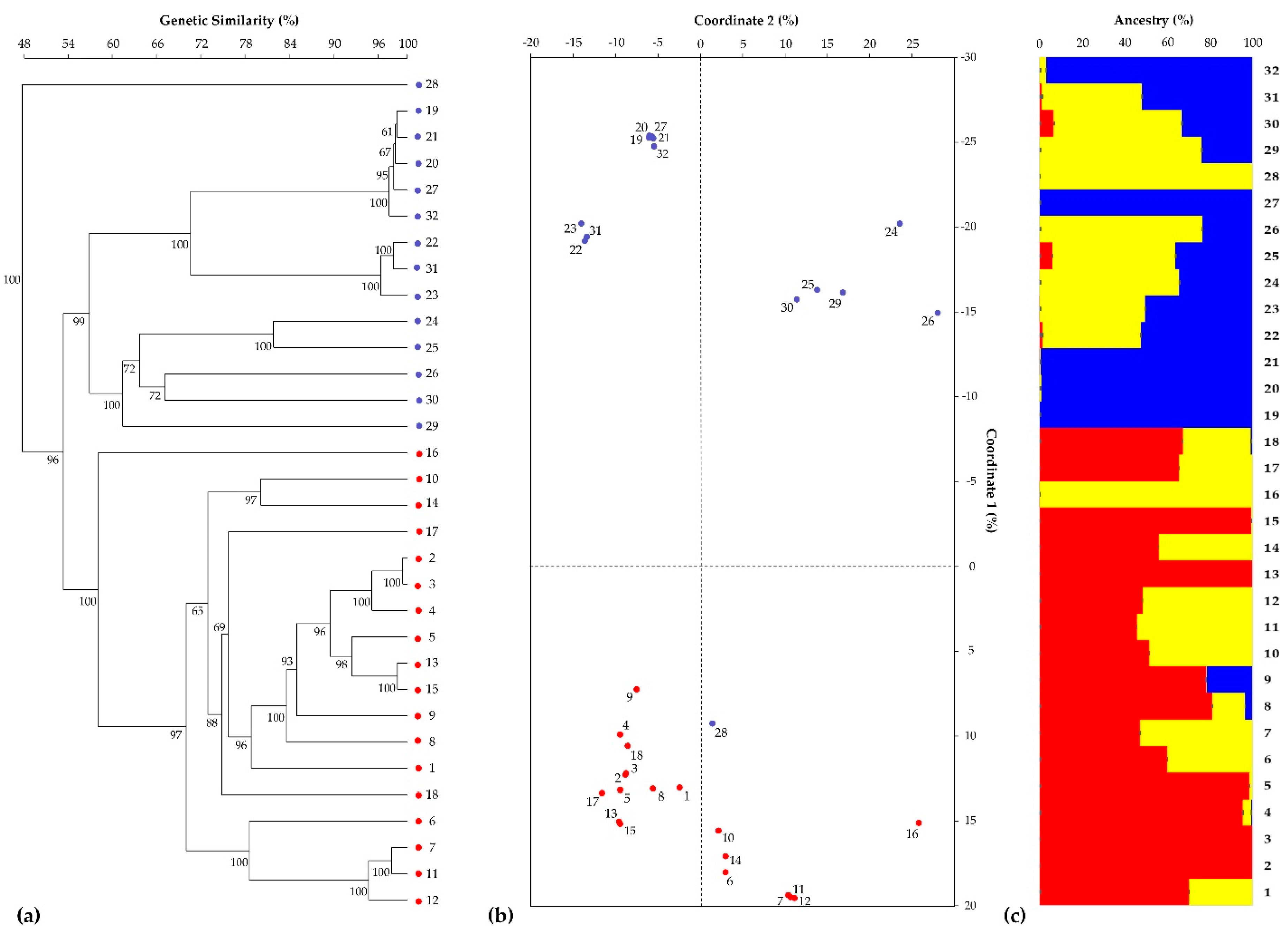
3.1. SSR-Based Genotyping by Heterologous Chicory-Derived Microsatellites
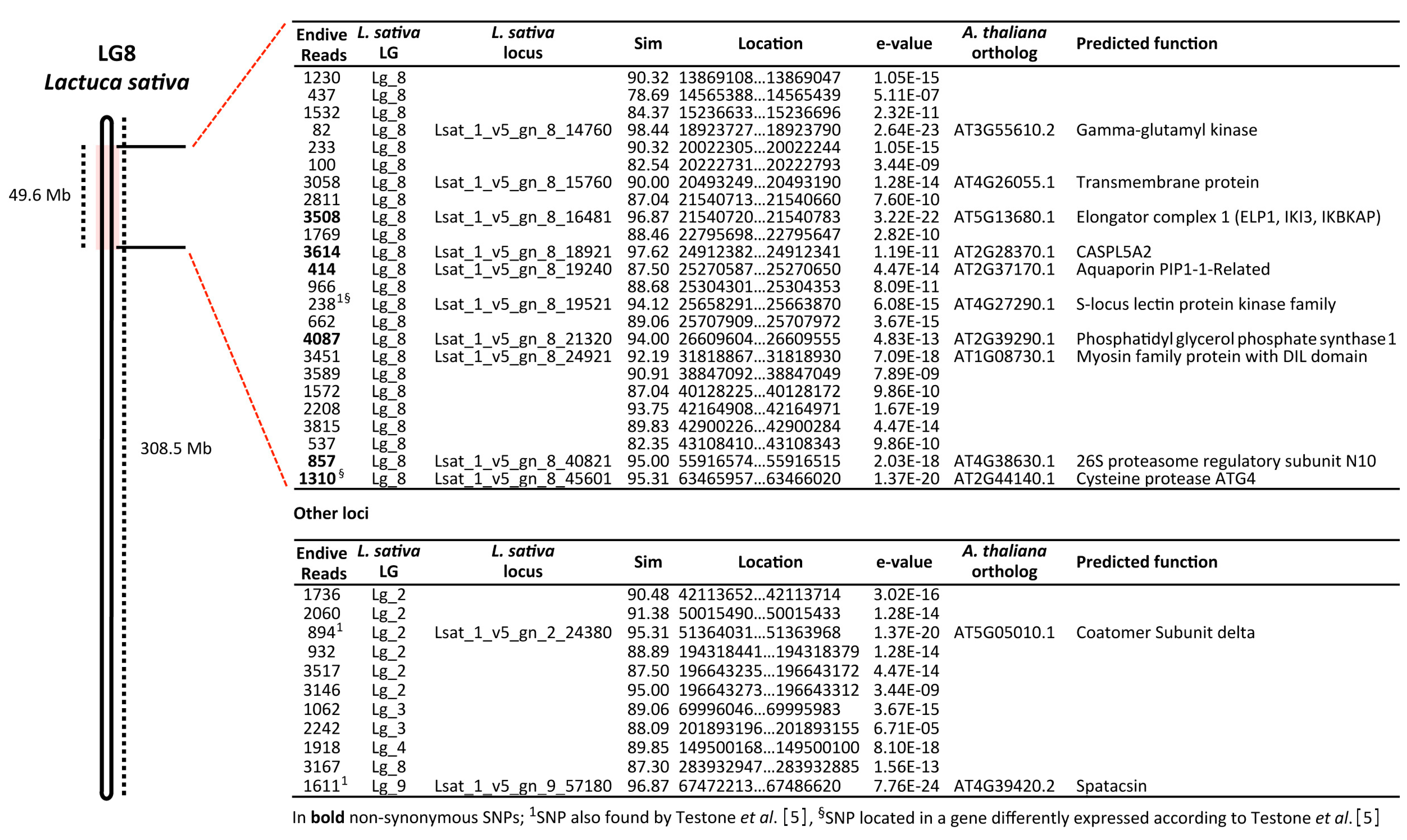
3.2. SNP-Based Genotyping by RAD-Seq Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Raulier, P.; Maudoux, O.; Notté, C.; Draye, X.; Bertin, P. Exploration of genetic diversity within Cichorium endivia and Cichorium intybus with focus on the gene pool of industrial chicory. Genet. Resour. Crop Evol. 2015, 63, 243–259. [Google Scholar] [CrossRef]
- Kowalczyk, K.; Gajc-Wolska, J.; Marcinkowska, M.; Jabrucka-Pióro, E. Assessment of quality attributes of endive (Cichorium endivia L.) depending on a cultivar and growing conditions. Acta Sci. Pol. Hortorum Cultus 2015, 14, 13–16. [Google Scholar]
- D’Antuono, L.F.; Ferioli, F.; Manco, M.A. The impact of sesquiterpene lactones and phenolics on sensory attributes: An investigation of a curly endive and escarole germplasm collection. Food Chem. 2016, 199, 238–245. [Google Scholar] [CrossRef]
- Lucchin, M.; Varotto, S.; Barcaccia, G.; Parrini, P. Chicory and endive. In Vegetables I; Springer: New York, NY, USA, 2008; pp. 3–48. [Google Scholar]
- Testone, G.; Mele, G.; di Giacomo, E.; Tenore, G.C.; Gonnella, M.; Nicolodi, C.; Frugis, G.; Iannelli, M.A.; Arnesi, G.; Schiappa, A.; et al. Transcriptome driven characterization of curly- and smooth-leafed endives reveals molecular differences in the sesquiterpenoid pathway. Hortic. Res. 2019, 6, 1–19. [Google Scholar] [CrossRef]
- Wang, F.-X.; Deng, A.-J.; Li, M.; Wei, J.-F.; Qin, H.-L.; Wang, A.-P. (3S)-1,2,3,4-Tetrahydro-β-carboline-3-carboxylic Acid from Cichorium endivia. L Induces Apoptosis of Human Colorectal Cancer HCT-8 Cells. Molecules 2012, 18, 418–429. [Google Scholar] [CrossRef]
- Wang, S.Z.; Wang, B.C.; Liu, J.; Ren, J.; Huang, X.X.; Zhou, G.L.; Wang, A.H. Novel polymorphic EST-based microsatellite markers characterized in lettuce (Lactuca sativa). Biologia 2017, 72, 1300–1305. [Google Scholar] [CrossRef]
- Ryder, E. Physiology of germination, growth and development. In Lettuce, Endive Chicory. Crop Production Science in Horticulture; Cabi Publishing: Wallingford, UK, 1999; pp. 54–78. [Google Scholar]
- Van Inghelandt, D.; Melchinger, A.E.; Lebreton, C.; Stich, B. Population structure and genetic diversity in a commercial maize breeding program assessed with SSR and SNP markers. Theor. Appl. Genet. 2010, 120, 1289–1299. [Google Scholar] [CrossRef] [Green Version]
- Lawson, C. Plant Breeder’s Rights and Essentially Derived Varieties: Still Searching for Workable Solutions; European Intellectual Property Review 499; Griffith University Law School Research Paper No. 16-17; Griffith University: Mount Gravatt, Australia, 2016. [Google Scholar]
- Patella, A.; Palumbo, F.; Galla, G.; Barcaccia, G. The Molecular Determination of Hybridity and Homozygosity Estimates in Breeding Populations of Lettuce (Lactuca sativa L.). Genes 2019, 10, 916. [Google Scholar] [CrossRef] [Green Version]
- Galla, G.; Ghedina, A.; Tiozzo, S.C.; Barcaccia, G. Toward a First High-quality Genome Draft for Marker-assisted Breeding in Leaf Chicory, Radicchio (Cichorium intybus L.). In Plant Genomics; IntechOpen: Rijeka, Croatia, 2016; pp. 67–87. [Google Scholar]
- Barcaccia, G.; Pallottini, L.; Soattin, M.; Lazzarin, R.; Parrini, P.; Lucchin, M. Genomic DNA fingerprints as a tool for identifying cultivated types of radicchio (Cichorium intybus L.) from Veneto, Italy. Plant Breed. 2003, 122, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Ghedina, A.; Galla, G.; Cadalen, T.; Hilbert, J.L.; Caenazzo, S.T.; Barcaccia, G. A method for genotyping elite breeding stocks of leaf chicory (Cichorium intybus L.) by assaying mapped microsatellite marker loci. BMC Res. Notes 2015, 8, 831–843. [Google Scholar] [CrossRef] [Green Version]
- Patella, A.; Scariolo, F.; Palumbo, F.; Barcaccia, G. Genetic Structure of Cultivated Varieties of Radicchio (Cichorium intybus L.): A Comparison between F1 Hybrids and Synthetics. Plants 2019, 8, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palumbo, F.; Qi, P.; Pinto, V.B.; Devos, K.M.; Barcaccia, G. Construction of the First SNP-Based Linkage Map Using Genotyping-by-Sequencing and Mapping of the Male-Sterility Gene in Leaf Chicory. Front. Plant Sci. 2019, 10, 276. [Google Scholar] [CrossRef] [PubMed]
- Cadalen, T.; Morchen, M.; Blassiau, C.; Clabaut, A.; Scheer, I.; Hilbert, J.L.; Hendriks, T.; Quillet, M.C. Development of SSR markers and construction of a consensus genetic map for chicory (Cichorium intybus L.). Mol. Breed. 2010, 25, 699–722. [Google Scholar] [CrossRef]
- De Simone, M.; Morgante, M.; Lucchin, M.; Parrini, P.; Marocco, A.J.M.B. A first linkage map of Cichorium intybus L. using a one-way pseudo-testcross and PCR-derived markers. Mol. Breed. 1997, 3, 415–425. [Google Scholar] [CrossRef]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233–234. [Google Scholar] [CrossRef]
- Yeh, F.C.; Yang, R.-C.; Boyle, T.B.J.; Ye, Z.-H.; Mao, J.X. POPGENE, the user-friendly shareware for population genetic analysis. In Molecular Biology and Biotechnology Centre; POPGENE Version 1.31; University of Alberta: Edmonton, AB, Canada, 1999. [Google Scholar]
- Hammer, Ø.; Harper, D.A.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Earl, D.A.; Vonholdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Stevanato, P.; Broccanello, C.; Biscarini, F.; Del Corvo, M.; Sablok, G.; Panella, L.; Stella, A.; Concheri, G. High-Throughput RAD-SNP Genotyping for Characterization of Sugar Beet Genotypes. Plant Mol. Biol. Rep. 2014, 32, 691–696. [Google Scholar] [CrossRef]
- Rochette, N.C.; Rivera-Colon, A.G.; Catchen, J.M. Stacks 2: Analytical methods for paired-end sequencing improve RADseq-based population genomics. Mol. Ecol. 2019, 28, 4737–4754. [Google Scholar] [CrossRef]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Reyes-Chin-Wo, S.; Wang, Z.; Yang, X.; Kozik, A.; Arikit, S.; Song, C.; Xia, L.; Froenicke, L.; Lavelle, D.O.; Truco, M.J.; et al. Genome assembly with in vitro proximity ligation data and whole-genome triplication in lettuce. Nat. Commun. 2017, 8, 14953. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314. [Google Scholar]
- Hoshino, A.A.; Bravo, J.P.; Angelici, C.M.L.C.D.; Barbosa, A.V.G.; Lopes, C.R.; Gimenes, M.A. Heterologous microsatellite primer pairs informative for the whole genus Arachis. Genet. Mol. Biol. 2006, 29, 665–675. [Google Scholar] [CrossRef]
- Wang, M.; Gillaspie, A.; Newman, M.; Dean, R.; Pittman, R.; Morris, J.; Pederson, G. Transfer of simple sequence repeat (SSR) markers across the legume family for germplasm characterization and evaluation. Plant Genet. Resour. Charact. Util. 2004, 2, 107–119. [Google Scholar] [CrossRef]
- Kalia, R.K.; Rai, M.K.; Kalia, S.; Singh, R.; Dhawan, A.K. Microsatellite markers: An overview of the recent progress in plants. Euphytica 2011, 177, 309–334. [Google Scholar] [CrossRef]
- Barbara, T.; Palma-Silva, C.; Paggi, G.M.; Bered, F.; Fay, M.F.; Lexer, C. Cross-species transfer of nuclear microsatellite markers: Potential and limitations. Mol. Ecol. 2007, 16, 3759–3767. [Google Scholar] [CrossRef]
- Bombonato, J.R.; Bonatelli, I.A.S.; Silva, G.A.R.; Moraes, E.M.; Zappi, D.C.; Taylor, N.P.; Franco, F.F. Cross-genera SSR transferability in cacti revealed by a case study using Cereus (Cereeae, Cactaceae). Genet. Mol. Biol. 2019, 42, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Aiello, D.; Ferradini, N.; Torelli, L.; Volpi, C.; Lambalk, J.; Russi, L.; Albertini, E. Evaluation of Cross-Species Transferability of SSR Markers in Foeniculum vulgare. Plants 2020, 9, 175. [Google Scholar] [CrossRef] [Green Version]
- Manju, K.P.; Manimekalai, R.; Naganeeswaran, S.A.; Arunachalam, V.; Karun, A. Microsatellites mining in date palm (Phoenix dactylifera L.) and their cross transferability across ‘Arecaceae’ family. Plant Omics 2016, 9, 191. [Google Scholar] [CrossRef]
- Buso, G.; Reis, A.; Amaral, Z.; Ferreira, M. Novel and highly informative Capsicum SSR markers and their cross-species transferability. Genet. Mol. Res. 2016, 15, 1–13. [Google Scholar] [CrossRef]
- Alexander, L.W.; Thammina, C.S.; Kramer, M. Cross-transferability of SSR markers in Osmanthus. Genet. Resour. Crop Evol. 2018, 65, 125–136. [Google Scholar] [CrossRef]
- Bazzo, B.R.; de Carvalho, L.M.; Carazzolle, M.F.; Pereira, G.A.G.; Colombo, C.A. Development of novel EST-SSR markers in the macauba palm (Acrocomia aculeata) using transcriptome sequencing and cross-species transferability in Arecaceae species. BMC Plant Biol. 2018, 18, 276. [Google Scholar] [CrossRef]
- Kim, J.M.; Lyu, J.I.; Lee, M.-K.; Kim, D.-G.; Kim, J.-B.; Ha, B.-K.; Ahn, J.-W.; Kwon, S.-J. Cross-species transferability of EST-SSR markers derived from the transcriptome of kenaf (Hibiscus cannabinus L.) and their application to genus Hibiscus. Genet. Resour. Crop Evol. 2019, 66, 1543–1556. [Google Scholar] [CrossRef]
- Kiers, A.M.; Mes, T.H.M.; Van der Meijden, R.; Bachmann, K. Morphologically defined Cichorium (Asteraceae) species reflect lineages based on chloroplast and nuclear (ITS) DNA data. Syst. Bot. 1999, 24, 645–659. [Google Scholar] [CrossRef]
- Kim, C.; Guo, H.; Kong, W.; Chandnani, R.; Shuang, L.S.; Paterson, A.H. Application of genotyping by sequencing technology to a variety of crop breeding programs. Plant Sci. 2016, 242, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhang, Z.; Yan, P.; Huang, S.; Fei, Z.; Lin, K. RNA-Seq improves annotation of protein-coding genes in the cucumber genome. BMC Genom. 2011, 12, 540. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.Y.; Li, M.; Zhao, S.; Zhang, C.; Yang, S.T.; Qiao, S.; Tan, W.F.; Qu, H.J.; Wang, D.Y.; Pu, Z.G. Analysis of evolution and genetic diversity of sweetpotato and its related different polyploidy wild species I. trifida using RAD-seq. BMC Plant Biol. 2018, 18, 181. [Google Scholar] [CrossRef]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 2016, 17, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Michael, T.P. Plant genome size variation: Bloating and purging DNA. Brief. Funct. Genom. 2014, 13, 308–317. [Google Scholar] [CrossRef]
- Zhang, Y.; Fan, X.C.; Li, Y.F.; Sun, H.S.; Jiang, J.F.; Liu, C.H. Restriction site-associated DNA sequencing reveals the molecular genetic diversity of grapevine and genes related to white rot disease. Sci. Hortic. 2020, 261, 108907. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Z.L.; Du, X.F.; Yang, H.Q.; Han, F.; Han, Y.H.; Yuan, F.; Zhang, L.Y.; Peng, S.Z.; Guo, E.H. A high-density genetic map and QTL analysis of agronomic traits in foxtail millet [Setaria italica (L.) P. Beauv.] using RAD-seq. PLoS ONE 2017, 12, e0179717. [Google Scholar] [CrossRef]
- Buckley, J.; Holub, E.B.; Koch, M.A.; Vergeer, P.; Mable, B.K. Restriction associated DNA-genotyping at multiple spatial scales in Arabidopsis lyrata reveals signatures of pathogen-mediated selection. BMC Genom. 2018, 19, 496. [Google Scholar] [CrossRef]
- Jaroslav, D.; Šimkova, H.; Kubaláková, M.; Šafář, J.; Suchánková, P.; Číhalíková, J.; Bartoš, J.; Valárik, M. Chromosome Genomics in the Triticeae. In Genetics and Genomics of the Triticeae, Plant Genetics and Genomics: Crops and Models 7; Feuillet, C., Muehlbauer, M.G., Eds.; Springer: New York, NY, USA, 2009; pp. 285–316. [Google Scholar] [CrossRef]
- Das, A.; Saxena, S.; Kumar, K.; Tribhuvan, K.U.; Singh, N.K.; Gaikwad, K. Non-coding RNAs having strong positive interaction with mRNAs reveal their regulatory nature during flowering in a wild relative of pigeonpea (Cajanus scarabaeoides). Mol. Biol. Rep. 2020. [Google Scholar] [CrossRef]
- Pootakham, W.; Sonthirod, C.; Naktang, C.; Jomchai, N.; Sangsrakru, D.; Tangphatsornruang, S. Effects of methylation-sensitive enzymes on the enrichment of genic SNPs and the degree of genome complexity reduction in a two-enzyme genotyping-by-sequencing (GBS) approach: A case study in oil palm (Elaeis guineensis). Mol. Breed. 2016, 36, 154. [Google Scholar] [CrossRef] [Green Version]
- Queirós, J.; Godinho, R.; Lopes, S.; Gortazar, C.; De la Fuente, J.; Alves, P. Effect of microsatellite selection on individual and population genetic inferences: An empirical study using cross-specific and species-specific amplifications. Mol. Ecol. Resour. 2015, 15, 747–760. [Google Scholar] [CrossRef]
- Cortés-Olmos, C.; Vilanova, S.; Pascual, L.; Rosello, J.; Cebolla-Cornejo, J. SNP markers applied to the characterization of Spanish tomato (Solanum lycopersicum L.) landraces. Sci. Hortic. 2015, 194, 100–110. [Google Scholar] [CrossRef]
- Verwaaijen, B.; Wibberg, D.; Nelkner, J.; Gordin, M.; Rupp, O.; Winkler, A.; Bremges, A.; Blom, J.; Grosch, R.; Puhler, A.; et al. Assembly of the Lactuca sativa, L. cv. Tizian draft genome sequence reveals differences within major resistance complex 1 as compared to the cv. Salinas reference genome. J. Biotechnol. 2018, 267, 12–18. [Google Scholar] [CrossRef]
- Schmidt, R.; Kunkowska, A.B.; Schippers, J.H. Role of Reactive Oxygen Species during Cell Expansion in Leaves. Plant Physiol. 2016, 172, 2098–2106. [Google Scholar] [CrossRef] [Green Version]
- Roppolo, D.; Boeckmann, B.; Pfister, A.; Boutet, E.; Rubio, M.C.; Denervaud-Tendon, V.; Vermeer, J.E.; Gheyselinck, J.; Xenarios, I.; Geldner, N. Functional and Evolutionary Analysis of the CASPARIAN STRIP MEMBRANE DOMAIN PROTEIN Family. Plant Physiol. 2014, 165, 1709–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woloszynska, M.; Gagliardi, O.; Vandenbussche, F.; Van Lijsebettens, M. Elongator promotes germination and early post-germination growth. Plant Signal. Behav. 2018, 13, e1422465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, C.; Wang, C.; Mou, Z. The Arabidopsis Elongator complex is required for nonhost resistance against the bacterial pathogens Xanthomonas citri subsp. citri and Pseudomonas syringae pv. phaseolicola NPS 3121. New Phytol. 2017, 214, 1245–1259. [Google Scholar]
- An, C.; Ding, Y.; Zhang, X.; Wang, C.; Mou, Z. Elongator Plays a Positive Role in Exogenous NAD-Induced Defense Responses in Arabidopsis. Mol. Plant Microbe Interact. 2016, 29, 396–404. [Google Scholar] [CrossRef]
- DeFraia, C.T.; Zhang, X.D.; Mou, Z.L. Elongator subunit 2 is an accelerator of immune responses in Arabidopsis thaliana. Plant J. 2010, 64, 511–523. [Google Scholar] [CrossRef]
- Kojima, S.; Iwasaki, M.; Takahashi, H.; Imai, T.; Matsumura, Y.; Fleury, D.; Van Lijsebettens, M.; Machida, Y.; Machida, C. Asymmetric leaves2 and Elongator, a histone acetyltransferase complex, mediate the establishment of polarity in leaves of Arabidopsis thaliana. Plant Cell Physiol. 2011, 52, 1259–1273. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| ID | LG | Motif | Primer Sequence and Tail | Multiplex | |
|---|---|---|---|---|---|
| M2.4 | 2 | (GA)25 | F | [PAN3]CCAACGGATACCAAGGTGTT | 1 |
| R | AACCGCACGGGTTCTATG | ||||
| M2.5 | 2 | (CT)5CC(CT)13 TT(CT)5 | F | [PAN1]GTGCCGGTCTTCAGGTTACA | 1 |
| R | CGCCTACCGATTACGATTGA | ||||
| M3.7 | 3 | (CT)22 | F | TTCGAGTCTTGCCTTAATTGTT | 1 |
| R | [PAN1]CAGACGACCTTACGGCAACT | ||||
| M4.10a | 4 | (CT)22 | F | [PAN2]CATCACCTTCACGAAAAGCA | 1 |
| R | CGAAGACCATCCATCACCA | ||||
| M4.11a | 4 | (CT)12N5(CA)11 | F | [PAN3]GAAGGAACCTATGAACCAACCACTCA | 1 |
| R | GTTTTGAGCCTGAGCCAGA | ||||
| M1.3 | 1 | (CT)17 | F | [PAN3]TGGAGAAAAATGAAGCAC | 2 |
| R | GAATGAGTGAGAGAATGATAGGG | ||||
| M5.13 | 5 | (CT)23 | F | [M13]AGGCATAAAGAGGTGTGG | 2 |
| R | TCAAACATGAAAACCGCTC | ||||
| M6.17 | 6 | (CA)8(CT)18 | F | CGTGTCCAAACGCAAACATTAT | 2 |
| R | [PAN2]GCACAATTTTCCTACCACTTATCC | ||||
| M5.14 | 5 | (TC)11 | F | [M13]AAAGTCACACATCGCATTTCCT | 2 |
| R | GTAGCAGCAGCAGCCATCTT | ||||
| M4.11b | 4 | (TG)5CG(TG)7 | F | [M13]GCCATTCCTTTCAAGAGCAG | 2 |
| R | AACCCAAAACCGCAACAATA | ||||
| M3.9 | 3 | (CA)12 | F | CTGCTATGGACAGTTCCAGT | 3 |
| R | [PAN3]CAATTCAGTTGTGATAGACGC | ||||
| M7.20 | 7 | (CT)31 | F | [PAN2]ACACTCACTCACACTCCGTAA | 3 |
| R | GTCATGATGGCGTAAAAGTC | ||||
| M6.18 | 6 | (CT)16 | F | [PAN3]CTCAACGAATGCTTTGGACA | 3 |
| R | CCTCGCGGTAGCTTATTGTT | ||||
| M2.6 | 2 | (CT)26 | F | GGAGCAGGTAGAGTCCCATC | 3 |
| R | [PAN1]CGTTTGAAAATTTATACCAAAATG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patella, A.; Palumbo, F.; Ravi, S.; Stevanato, P.; Barcaccia, G. Genotyping by RAD Sequencing Analysis Assessed the Genetic Distinctiveness of Experimental Lines and Narrowed down the Genomic Region Responsible for Leaf Shape in Endive (Cichorium endivia L.). Genes 2020, 11, 462. https://doi.org/10.3390/genes11040462
Patella A, Palumbo F, Ravi S, Stevanato P, Barcaccia G. Genotyping by RAD Sequencing Analysis Assessed the Genetic Distinctiveness of Experimental Lines and Narrowed down the Genomic Region Responsible for Leaf Shape in Endive (Cichorium endivia L.). Genes. 2020; 11(4):462. https://doi.org/10.3390/genes11040462
Chicago/Turabian StylePatella, Alice, Fabio Palumbo, Samathmika Ravi, Piergiorgio Stevanato, and Gianni Barcaccia. 2020. "Genotyping by RAD Sequencing Analysis Assessed the Genetic Distinctiveness of Experimental Lines and Narrowed down the Genomic Region Responsible for Leaf Shape in Endive (Cichorium endivia L.)" Genes 11, no. 4: 462. https://doi.org/10.3390/genes11040462
APA StylePatella, A., Palumbo, F., Ravi, S., Stevanato, P., & Barcaccia, G. (2020). Genotyping by RAD Sequencing Analysis Assessed the Genetic Distinctiveness of Experimental Lines and Narrowed down the Genomic Region Responsible for Leaf Shape in Endive (Cichorium endivia L.). Genes, 11(4), 462. https://doi.org/10.3390/genes11040462