Comparative Transcriptomic Analysis to Identify the Genes Related to Delayed Gland Morphogenesis in Gossypium bickii


 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. RNA Extraction, Library Construction and RNA-Seq Analysis
2.2. Analysis of Differentially-Expressed Genes
2.3. Quantification, Gene Ontology and KEGG Pathway in Differential Gene Expression
2.4. Construction of Gene Network
2.5. qRT-PCR Analysis to Validate RNA-Seq Data
3. Results
3.1. Summary of Transcriptome Data
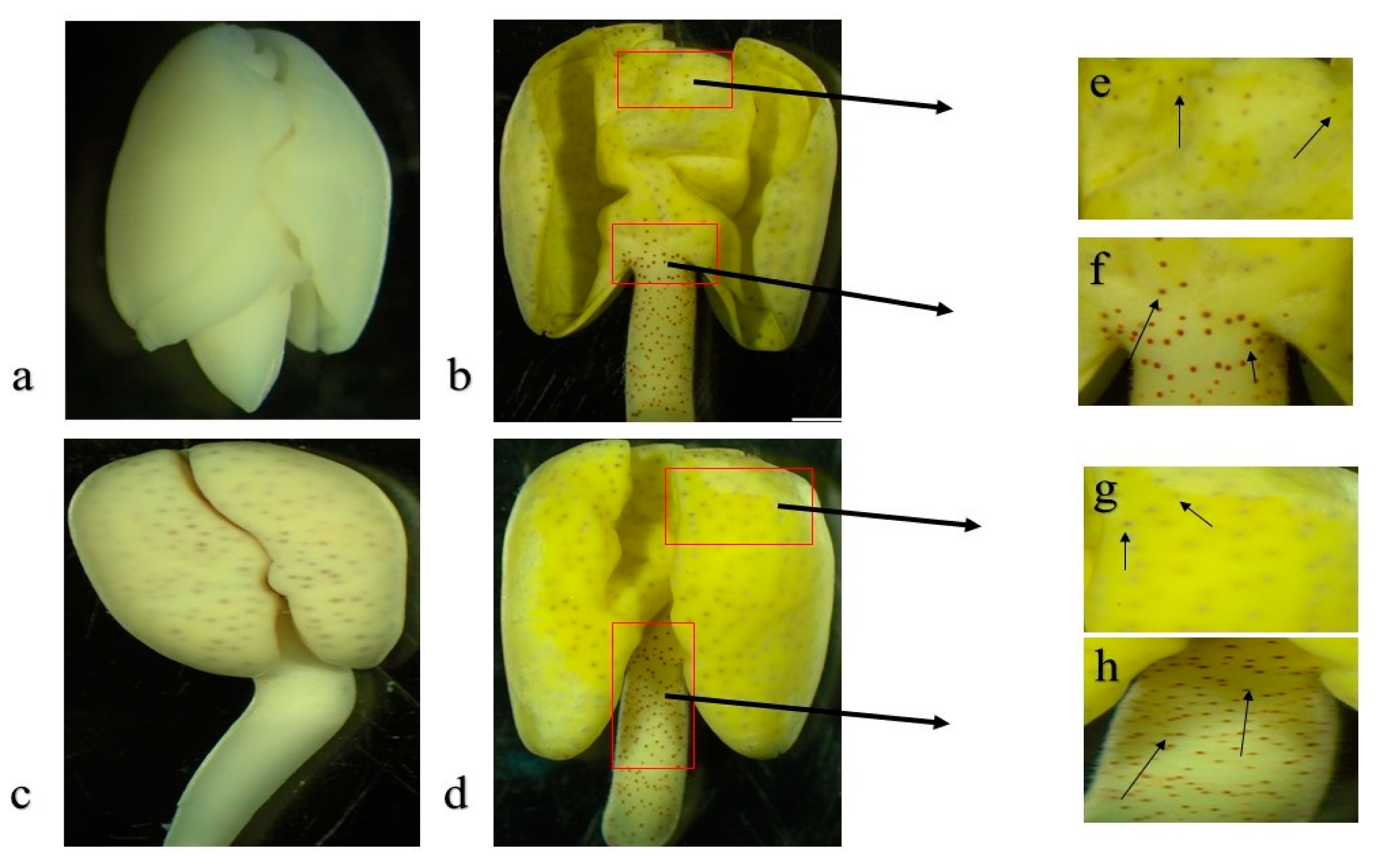
3.2. Transcriptome Changes during Imbibed Seed (Glandless) and Seedling (Glanded Stages)
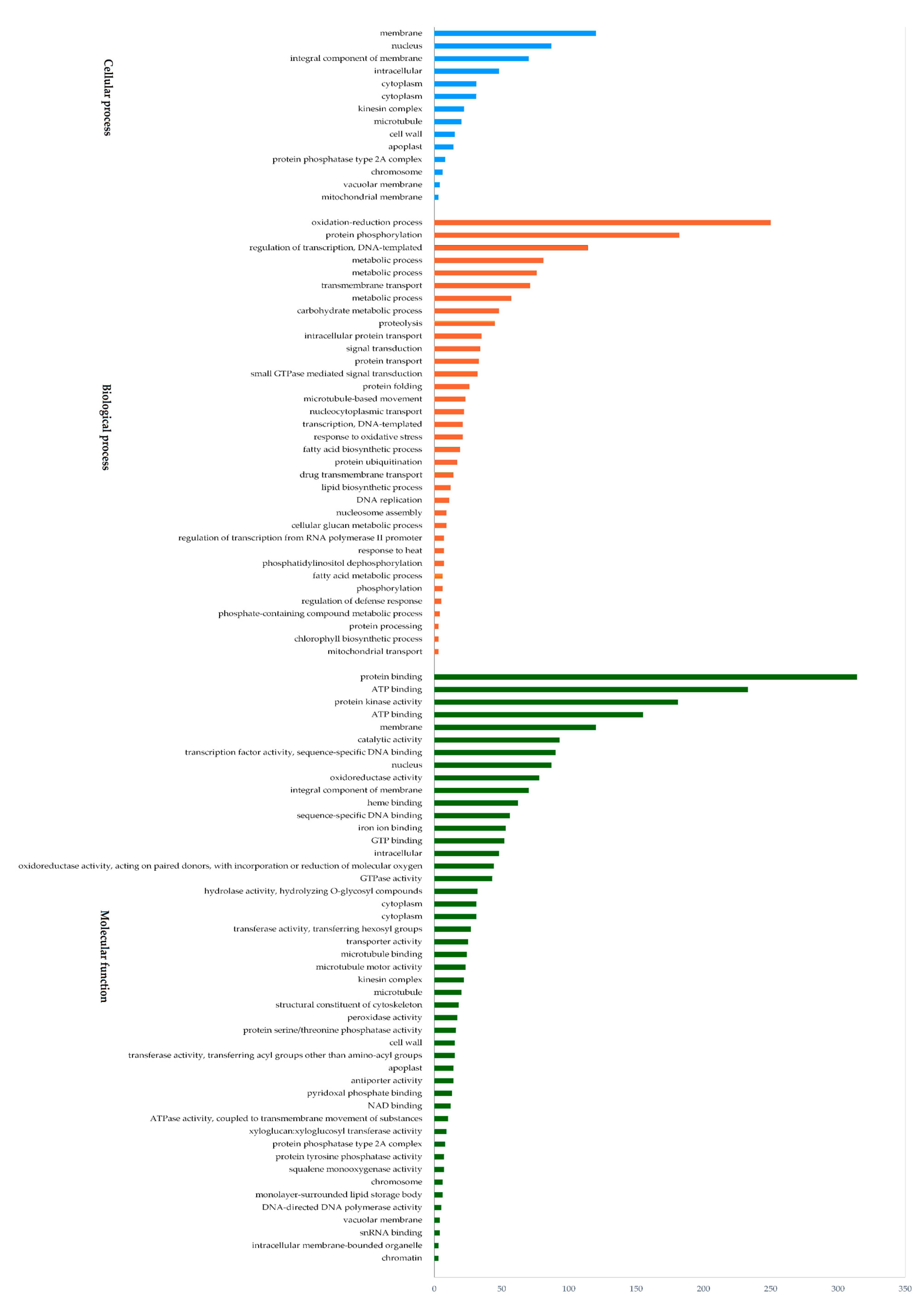
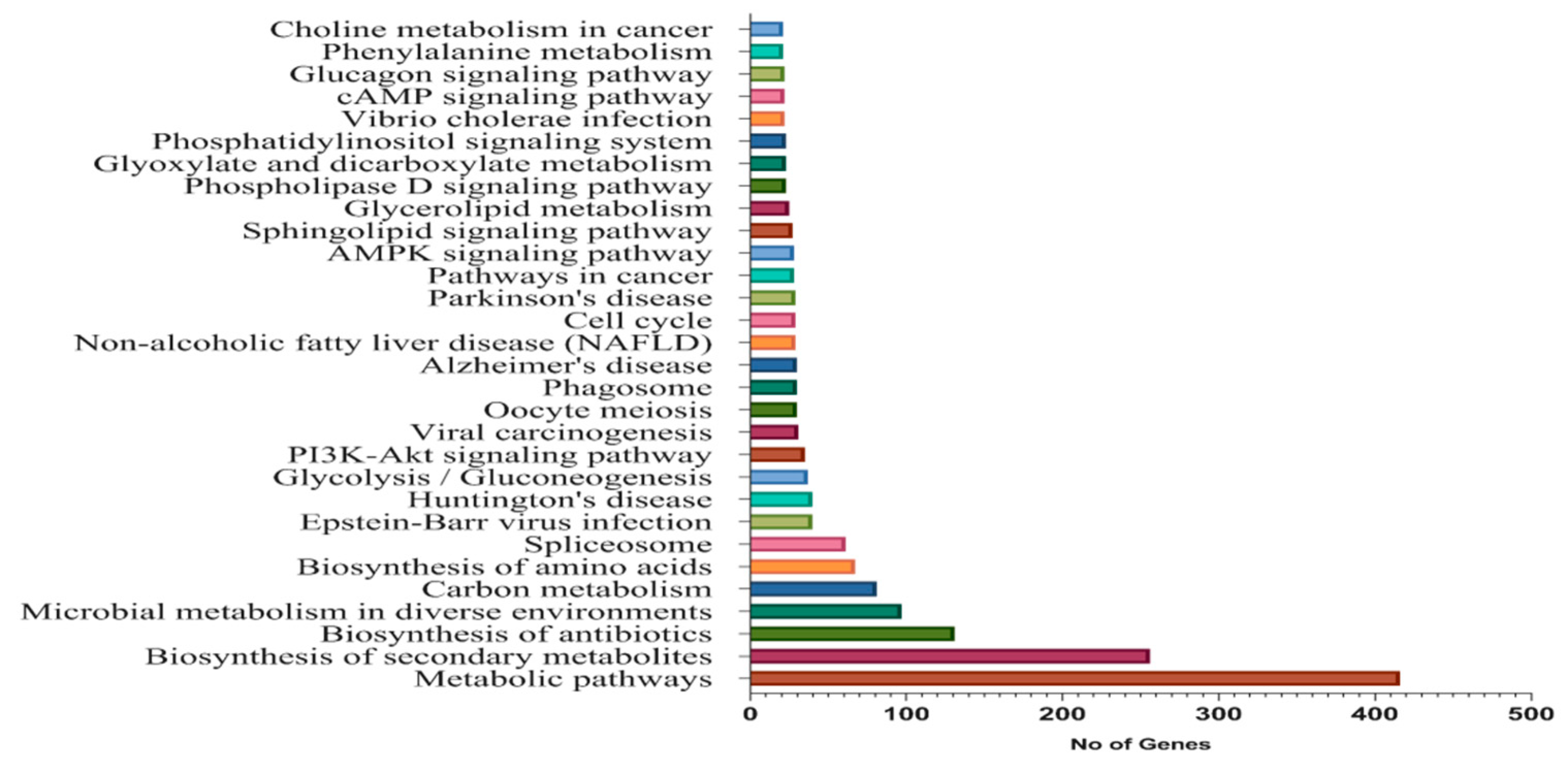
3.3. Functional Annotations of Differentially-Expressed Genes
3.4. Gene Co-Expression Correlation Network Analysis
3.5. Quantitative Real-Time PCR Validation of RNA-Seq Data
4. Discussion
4.1. Comparison of Expression Profiles, RNA Sequences between Glandless G. bickii and Glanded G. arboreum
4.2. Hub Genes Identified Using WGCNA
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Gbgl | Gossypium bickii glandless imbibed seed |
| Gbdd | Gossypium bickii glanded seedlings |
| Ga24h | Gossypium arboreum glanded imbibed seed |
| Ga48h | Gossypium arboreum glanded seedlings |
| ME | Module Eigengenes |
References
- Ma, D.; Hu, Y.; Yang, C.; Liu, B.; Fang, L.; Wan, Q.; Liang, W.; Mei, G.; Wang, L.; Wang, H. Genetic basis for glandular trichome formation in cotton. Nat. Commun. 2016, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Robert-Seilaniantz, A.; Grant, M.; Jones, J.D. Hormone crosstalk in plant disease and defense: More than just jasmonate-salicylate antagonism. Ann. Rev. Phytopathol. 2011, 49, 317–343. [Google Scholar] [CrossRef] [PubMed]
- Fryxell, P.A. A redefinition of the tribe Gossypieae. Bot. Gaz. 1968, 129, 296–308. [Google Scholar] [CrossRef]
- Tian, X.; Ruan, J.-X.; Huang, J.-Q.; Yang, C.-Q.; Fang, X.; Chen, Z.-W.; Hong, H.; Wang, L.-J.; Mao, Y.-B.; Lu, S. Characterization of gossypol biosynthetic pathway. Proc. Natl. Acad. Sci. USA 2018, 115, E5410–E5418. [Google Scholar] [CrossRef] [Green Version]
- Stanford, E.E.; Viehoever, A. Chemistry and histology of the glands of the cotton plant, with notes on the occurrence of similar glands in related plants. J. Agric. Res 1918, 13, 419–435. [Google Scholar]
- Maxwell, W.D. The Liturgical Portions of the Genevan Service Book, Used by John Knox While a Minister of the English Congregation of Marian Exiles at Geneva, Switzerland 1556-9. John Knox’s Genevan Service Book, 1556; Faith Press: Geneva, Switezerland, 1965. [Google Scholar]
- Lukefahr, M.; Martin, D. Cotton-plant pigments as a source of resistance to the bollworm and tobacco budworm. J. Econ. Entomol. 1966, 59, 176–179. [Google Scholar] [CrossRef]
- Stipanovic, R.D.; Bell, A.A.; O’Brien, D.H.; Lukefahr, M.J. Heliocide H1. A new insecticidal C25 terpenoid from cotton (Gossypium hirsutum). J. Agric. Food Chem. 1978, 26, 115–118. [Google Scholar] [CrossRef]
- McMichael, S.C. Hopi cotton, a source of cottonseed free of gossypol pigments 1. Agron. J. 1959, 51, 630. [Google Scholar] [CrossRef]
- McMichael, S.C. Combined effects of glandless genes gl2 and gl3 on pigment glands in the cotton plant. Agron. J. 1960, 52, 385–386. [Google Scholar] [CrossRef]
- Miravalle, R.J.; Hyer, A.H. Identification of the Gl2 gl2 Gl3 gl3 genotype in breeding for glandless cottonseed 1. Crop Sci. 1962, 2, 395–397. [Google Scholar] [CrossRef]
- Lee, J.A. The genomic allocation of the principal foliar-gland loci in Gossypium hirsutum and Gossypium barbadense. Evolution 1965, 19, 182–188. [Google Scholar] [CrossRef]
- Murray, R. The aetiology of primary osteoarthritis of the hip. Br. J. Radiol. 1965, 38, 810–824. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.K.; Tang, C.; Chen, Y.-L.; Wu, C.-W. Nuclear proteins of the bovine esophageal epithelium. II. The NuMA gene gives rise to multiple mRNAs and gene products reactive with monoclonal antibody W1. J. Cell Sci. 1993, 104, 249–260. [Google Scholar] [PubMed]
- Xianhe, J.S.Z. Selection of new types of dominant glandless cotton (Gossypium hirsutum) germplasm. Sci. Agric. Sin. 1990, 4, 22–27. [Google Scholar]
- Wilkins, T.A.; Arpat, A.; Sickler, B. Cotton fiber genomics: Developmental mechanisms. Pflanzenschutz-Nachrichten Bayer 2005, 58, 119–139. [Google Scholar]
- Al-Ghazi, Y.; Bourot, S.; Arioli, T.; Dennis, E.S.; Llewellyn, D.J. Transcript profiling during fiber development identifies pathways in secondary metabolism and cell wall structure that may contribute to cotton fiber quality. Plant Cell Physiol. 2009, 50, 1364–1381. [Google Scholar] [CrossRef]
- Arpat, A.; Waugh, M.; Sullivan, J.P.; Gonzales, M.; Frisch, D.; Main, D.; Wood, T.; Leslie, A.; Wing, R.; Wilkins, T. Functional genomics of cell elongation in developing cotton fibers. Plant Mol. Biol. 2004, 54, 911–929. [Google Scholar] [CrossRef]
- Li, P.-t.; Wang, M.; Lu, Q.-w.; Ge, Q.; Liu, A.-y.; Gong, J.-w.; Shang, H.-h.; Gong, W.-k.; Li, J.-w.; Song, W.-w. Comparative transcriptome analysis of cotton fiber development of Upland cotton (Gossypium hirsutum) and chromosome segment substitution lines from G. hirsutum × G. barbadense. BMC Genom. 2017, 18, 705. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.; Liu, A.; Zhang, Z.; Ge, Q.; Fan, S.; Gong, W.; Li, J.; Gong, J.; Shi, Y.; Tian, B. Co-expression network analysis and hub gene selection for high-quality fiber in upland cotton (Gossypium hirsutum) using RNA sequencing analysis. Genes 2019, 10, 119. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Qin, T.; Wei, C.; Sun, J.; Dong, T.; Zhou, R.; Chen, Q.; Wang, Q. Using transcriptome analysis to screen for key genes and pathways related to cytoplasmic male sterility in cotton (Gossypium hirsutum L.). Int. J. Mol. Sci. 2019, 20, 5120. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Lu, C.; John, Z.Y.; Zou, C.; Zhang, Y.; Wang, Q.; Huang, J.; Feng, X.; Jiang, P.; Yang, W. Fine mapping and candidate gene analysis of the dominant glandless gene Gl 2 e in cotton (Gossypium spp.). Theor. Appl. Genet. 2016, 129, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Cai, X.; Wang, Q.; Wang, P.; Zhang, Y.; Cai, C.; Xu, Y.; Wang, K.; Zhou, Z.; Wang, C. Genome sequencing of the Australian wild diploid species Gossypium australe highlights disease resistance and delayed gland morphogenesis. Plant Biotechnol. J. 2019, 18, 814–828. [Google Scholar] [CrossRef] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511. [Google Scholar] [CrossRef] [Green Version]
- Talon, M.; Gmitter, F.G. Citrus genomics. Int. J. Plant Genom. 2008, 2008, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 4. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Livak, K.; Schmittgen, T. Analysis of relative gene expression data using real-time quantitative PCR and the 2-DDCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Yang, Y.; Bao, S.; Zhou, X.; Liu, J.; Zhuang, Y. The key genes and pathways related to male sterility of eggplant revealed by comparative transcriptome analysis. BMC Plant Biol. 2018, 18, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hollender, C.A.; Kang, C.; Darwish, O.; Geretz, A.; Matthews, B.F.; Slovin, J.; Alkharouf, N.; Liu, Z. Floral transcriptomes in woodland strawberry uncover developing receptacle and anther gene networks. Plant Physiol. 2014, 165, 1062–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Tu, L.; Yuan, D.; Zhu, D.; Shen, C.; Li, J.; Liu, F.; Pei, L.; Wang, P.; Zhao, G. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 2019, 51, 224–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 2012, 44, 1098–1103. [Google Scholar] [CrossRef]
- Li, F.; Fan, G.; Wang, K.; Sun, F.; Yuan, Y.; Song, G.; Li, Q.; Ma, Z.; Lu, C.; Zou, C. Genome sequence of the cultivated cotton Gossypium arboreum. Nat. Genet. 2014, 46, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Yoo, M.-J.; Wendel, J.F. Comparative evolutionary and developmental dynamics of the cotton (Gossypium hirsutum) fiber transcriptome. PLoS Genet. 2014, 10, e1004073. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.S.; Fang, D.D.; Thyssen, G.N.; Delhom, C.D.; Liu, Y.; Kim, H.J. Comparative fiber property and transcriptome analyses reveal key genes potentially related to high fiber strength in cotton (Gossypium hirsutum L.) line MD52ne. BMC Plant Biol. 2016, 16, 36. [Google Scholar] [CrossRef] [Green Version]
- Li, P.-T.; Chen, T.-T.; Lu, Q.-W.; Ge, Q.; Gong, W.-K.; Liu, A.-Y.; Gong, J.-W.; Shang, H.-H.; Deng, X.-Y.; Li, J.-W. Transcriptomic and biochemical analysis of upland cotton (Gossypium hirsutum) and a chromosome segment substitution line from G. hirsutum × G. barbadense in response to Verticillium dahliae infection. BMC Plant Biol. 2019, 19, 1–24. [Google Scholar] [CrossRef]
- Patel, R.; Baker, S.S.; Liu, W.; Desai, S.; Alkhouri, R.; Kozielski, R.; Mastrandrea, L.; Sarfraz, A.; Cai, W.; Vlassara, H. Effect of dietary advanced glycation end products on mouse liver. PLoS ONE 2012, 7, e35143. [Google Scholar] [CrossRef]
- Artico, S.; Ribeiro-Alves, M.; Oliveira-Neto, O.B.; de Macedo, L.L.P.; Silveira, S.; Grossi-de-Sa, M.F.; Martinelli, A.P.; Alves-Ferreira, M. Transcriptome analysis of Gossypium hirsutum flower buds infested by cotton boll weevil (Anthonomus grandis) larvae. BMC Genom. 2014, 15, 854. [Google Scholar] [CrossRef] [Green Version]
- Brubaker, C.; Benson, C.G.; Miller, C.; Leach, D.N. Occurrence of terpenoid aldehydes and lysigenous cavities in the’glandless’ seeds of Australian Gossypium species. Aust. J. Bot. 1996, 44, 601–612. [Google Scholar] [CrossRef]
- Zhu, S.; Ji, D. Inheritance of the delayed gland morphogenesis trait in Australian wild species of Gossypium. Chin. Sci. Bull. 2001, 46, 1168–1174. [Google Scholar] [CrossRef]
- Symonds, V.V.; Hatlestad, G.; Lloyd, A.M. Natural allelic variation defines a role for ATMYC1: Trichome cell fate determination. PLoS Genet. 2011, 7, e1002069. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Wang, X.; Zhu, D.; Cui, S.; Li, X.; Cao, Y.; Ma, L. A single amino acid substitution in IIIf subfamily of basic helix-loop-helix transcription factor AtMYC1 leads to trichome and root hair patterning defects by abolishing its interaction with partner proteins in Arabidopsis. J. Biol. Chem. 2012, 287, 14109–14121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, A.; Roychoudhury, A. WRKY proteins: Signaling and regulation of expression during abiotic stress responses. Sci. World J. 2015, 2015, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javelle, M.; Vernoud, V.; Rogowsky, P.M.; Ingram, G.C. Epidermis: The formation and functions of a fundamental plant tissue. New Phytol. 2011, 189, 17–39. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Raw Reads | Clean Reads | Clean Bases | Error Rate (%) | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|---|---|
| Gbdd_1 | 76,722,280 | 75,666,498 | 11.35G | 0.03 | 97.53 | 93.05 | 43.94 |
| Gbdd_2 | 68,409,156 | 67,453,570 | 10.12G | 0.03 | 97.63 | 93.31 | 43.63 |
| Gbdd_3 | 75,015,858 | 74,123,028 | 11.12G | 0.03 | 97.65 | 93.34 | 43.57 |
| Ga48h_1 | 70,605,862 | 69,735,728 | 10.46G | 0.03 | 97.45 | 92.81 | 43.91 |
| Ga48h_2 | 77,498,638 | 76,604,382 | 11.49G | 0.03 | 97.57 | 93.13 | 43.95 |
| Ga48h_3 | 76,083,930 | 75,133,632 | 11.27G | 0.03 | 97.37 | 92.64 | 43.77 |
| Gbgl_1 | 77,882,552 | 77,011,776 | 11.55G | 0.03 | 97.32 | 92.62 | 43.24 |
| Gbgl_2 | 66,734,644 | 65,760,564 | 9.86G | 0.03 | 97.2 | 92.36 | 43.25 |
| Gbgl_3 | 70,721,200 | 69,789,014 | 10.47G | 0.03 | 97.61 | 93.24 | 43.67 |
| Ga24h_1 | 70,796,734 | 69,833,972 | 10.48G | 0.03 | 97.62 | 93.21 | 43.04 |
| Ga24h_2 | 82,478,328 | 80,547,802 | 12.08G | 0.03 | 97.65 | 93.32 | 43.67 |
| Ga24h_3 | 74,698,432 | 73,859,056 | 11.08G | 0.03 | 97.17 | 92.17 | 43.25 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, M.; Cheng, H.; Soomro, M.; Shuyan, L.; Bilal Tufail, M.; Nazir, M.F.; Feng, X.; Zhang, Y.; Dongyun, Z.; Limin, L.; et al. Comparative Transcriptomic Analysis to Identify the Genes Related to Delayed Gland Morphogenesis in Gossypium bickii. Genes 2020, 11, 472. https://doi.org/10.3390/genes11050472
Ali M, Cheng H, Soomro M, Shuyan L, Bilal Tufail M, Nazir MF, Feng X, Zhang Y, Dongyun Z, Limin L, et al. Comparative Transcriptomic Analysis to Identify the Genes Related to Delayed Gland Morphogenesis in Gossypium bickii. Genes. 2020; 11(5):472. https://doi.org/10.3390/genes11050472
Chicago/Turabian StyleAli, Mushtaque, Hailiang Cheng, Mahtab Soomro, Li Shuyan, Muhammad Bilal Tufail, Mian Faisal Nazir, Xiaoxu Feng, Youping Zhang, Zuo Dongyun, Lv Limin, and et al. 2020. "Comparative Transcriptomic Analysis to Identify the Genes Related to Delayed Gland Morphogenesis in Gossypium bickii" Genes 11, no. 5: 472. https://doi.org/10.3390/genes11050472
APA StyleAli, M., Cheng, H., Soomro, M., Shuyan, L., Bilal Tufail, M., Nazir, M. F., Feng, X., Zhang, Y., Dongyun, Z., Limin, L., Wang, Q., & Song, G. (2020). Comparative Transcriptomic Analysis to Identify the Genes Related to Delayed Gland Morphogenesis in Gossypium bickii. Genes, 11(5), 472. https://doi.org/10.3390/genes11050472