Overcoming Immunological Challenges to Helper-Dependent Adenoviral Vector-Mediated Long-Term CFTR Expression in Mouse Airways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. HD-Ad Vector Preparation and Delivery to Mice Lungs
2.2. RNA Isolation and Real-Time RT-qPCR
2.3. Anti-Ad Antibody Titer Assay
2.4. Neutralizing Abs Assay
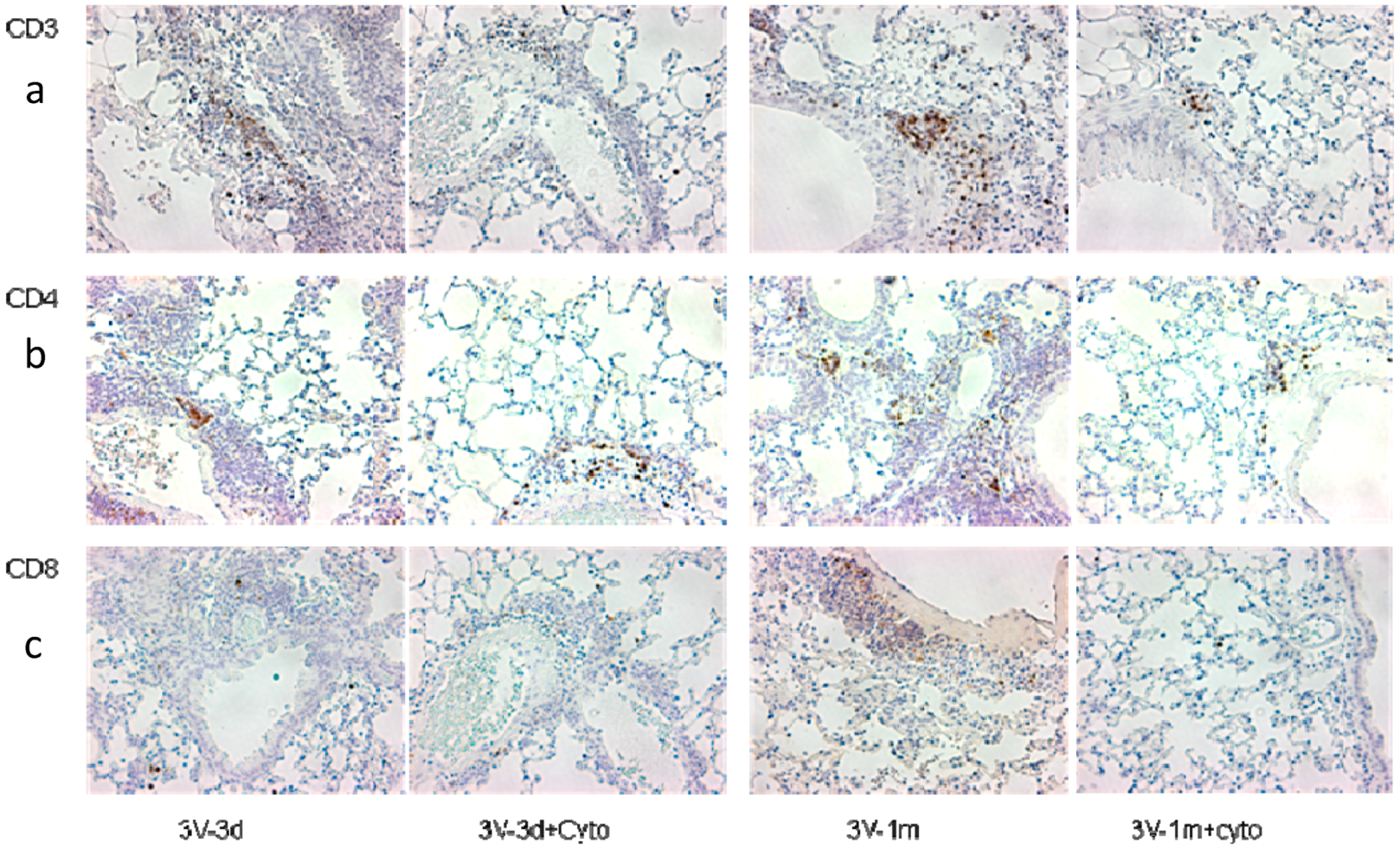
2.5. CDs Marker Staining
2.6. Statistical Analysis
3. Results
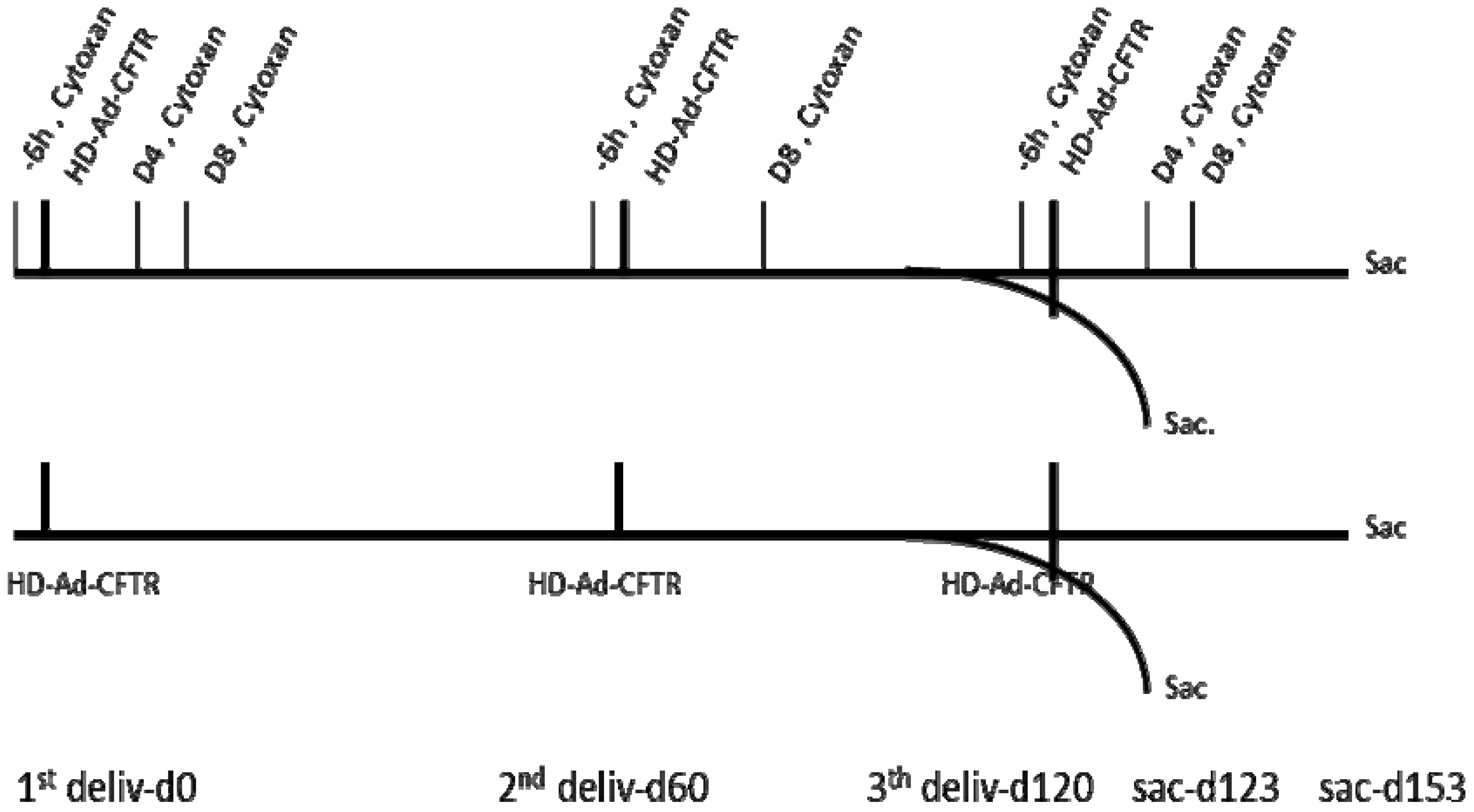
3.1. Scheme of Experiment Design for HD-Ad-CFTR Vector Delivery and Immunosuppressant Administration
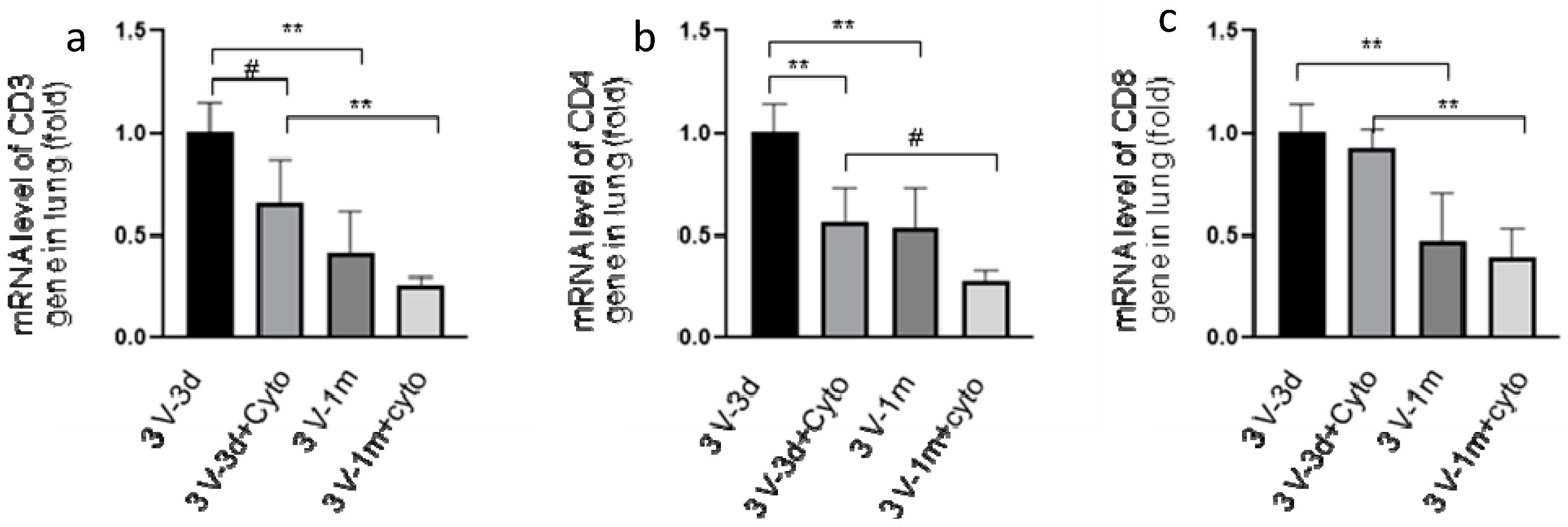
3.2. Cyclophosphamide Reduced T Cell Gene Expression
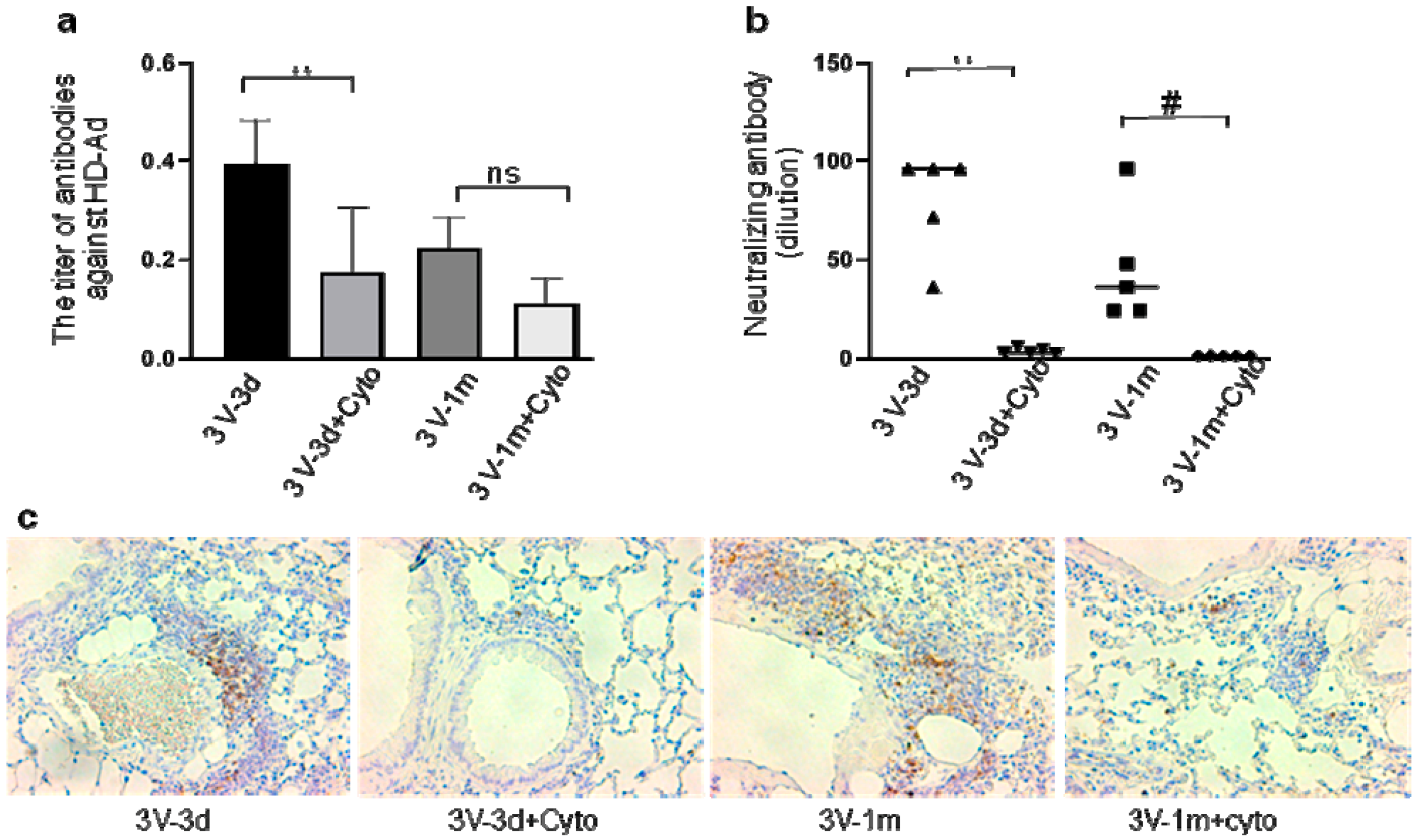
3.3. Cyclophosphamide Greatly Reduced B Cell Responses Induced by Repeated Vector Delivery
3.4. Transgene Expression was Improved Significantly by Immunosuppressant
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Corbyn, Z. Promising new era dawns for cystic fibrosis treatment. Lancet 2012, 379, 1475–1476. [Google Scholar] [CrossRef]
- Davies, J.C.; Moskowitz, S.M.; Brown, C.; Horsley, A.; Mall, M.A.; McKone, E.F.; Plant, B.J.; Prais, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; et al. VX-659-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1599–1611. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Cooney, A.L.; McCray, P.B., Jr.; Sinn, P.L. Cystic Fibrosis Gene Therapy: Looking Back, Looking Forward. Genes 2018, 9, 538. [Google Scholar] [CrossRef] [Green Version]
- Brody, H. Gene therapy. Nature 2018, 564, S5. [Google Scholar] [CrossRef]
- Jennings, M.T.; Flume, P.A. Cystic Fibrosis: Translating Molecular Mechanisms into Effective Therapies. Ann. Am. Thorac. Soc. 2018, 15, 897–902. [Google Scholar] [CrossRef]
- Ginn, S.L.; Amaya, A.K.; Alexander, I.E.; Edelstein, M.; Abedi, M.R. Gene therapy clinical trials worldwide to 2017: An update. J. Gene Med. 2018, 20, e3015. [Google Scholar] [CrossRef]
- Dunbar, C.E.; High, K.A.; Joung, J.K.; Kohn, D.B.; Ozawa, K.; Sadelain, M. Gene therapy comes of age. Science 2018, 359. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Mese, K.; Bunz, O.; Ehrhardt, A. State-of-the-art human adenovirus vectorology for therapeutic approaches. FEBS Lett. 2019, 593, 3609–3622. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Koehler, D.R.; Hu, J. Adenoviral vectors for gene replacement therapy. Viral Immunol. 2004, 17, 327–333. [Google Scholar] [CrossRef]
- Flotte, T.R.; Ng, P.; Dylla, D.E.; McCray, P.B., Jr.; Wang, G.; Kolls, J.K.; Hu, J. Viral vector-mediated and cell-based therapies for treatment of cystic fibrosis. Mol. Ther. 2007, 15, 229–241. [Google Scholar] [CrossRef]
- Koehler, D.R.; Hitt, M.M.; Hu, J. Challenges and strategies for cystic fibrosis lung gene therapy. Mol. Ther. 2001, 4, 84–91. [Google Scholar] [CrossRef]
- Zhou, Z.P.; Yang, L.L.; Cao, H.; Chen, Z.R.; Zhang, Y.; Wen, X.Y.; Hu, J. In Vitro Validation of a CRISPR-Mediated CFTR Correction Strategy for Preclinical Translation in Pigs. Hum. Gene Ther. 2019, 30, 1101–1116. [Google Scholar] [CrossRef]
- Xia, E.; Zhang, Y.; Cao, H.; Li, J.; Duan, R.; Hu, J. TALEN-Mediated Gene Targeting for Cystic Fibrosis-Gene Therapy. Genes 2019, 10, 39. [Google Scholar] [CrossRef] [Green Version]
- Xia, E.; Duan, R.; Shi, F.; Seigel, K.E.; Grasemann, H.; Hu, J. Overcoming the Undesirable CRISPR-Cas9 Expression in Gene Correction. Mol. Ther. Nucleic Acids 2018, 13, 699–709. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Machuca, T.N.; Yeung, J.C.; Wu, J.; Du, K.; Duan, C.; Hashimoto, K.; Linacre, V.; Coates, A.L.; Leung, K.; et al. Efficient gene delivery to pig airway epithelia and submucosal glands using helper-dependent adenoviral vectors. Mol. Ther. Nucleic Acids 2013, 2, e127. [Google Scholar] [CrossRef]
- Cao, H.; Yang, T.; Li, X.F.; Wu, J.; Duan, C.; Coates, A.L.; Hu, J. Readministration of helper-dependent adenoviral vectors to mouse airway mediated via transient immunosuppression. Gene Ther. 2011, 18, 173–181. [Google Scholar] [CrossRef]
- Cao, H.; Ouyang, H.; Grasemann, H.; Bartlett, C.; Du, K.; Duan, R.; Shi, F.; Estrada, M.; Seigel, K.; Coates, A.; et al. Transducing airway basal cells with a helper-dependent adenoviral vector for lung gene therapy. Hum. Gene Ther. 2018. [Google Scholar] [CrossRef]
- Levardon, H.; Yonker, L.M.; Hurley, B.P.; Mou, H. Expansion of Airway Basal Cells and Generation of Polarized Epithelium. Bio. Protoc. 2018, 8. [Google Scholar] [CrossRef]
- Carlon, M.S.; Vidovic, D.; Dooley, J.; da Cunha, M.M.; Maris, M.; Lampi, Y.; Toelen, J.; Van den Haute, C.; Baekelandt, V.; Deprest, J.; et al. Immunological ignorance allows long-term gene expression after perinatal recombinant adeno-associated virus-mediated gene transfer to murine airways. Hum. Gene Ther. 2014, 25, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, S.; Griesenbach, U.; Geddes, D.M.; Alton, E. Immunological hurdles to lung gene therapy. Clin. Exp. Immunol. 2003, 132, 1–8. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Tuddenham, E.G.; Rangarajan, S.; Rosales, C.; McIntosh, J.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C.; et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011, 365, 2357–2365. [Google Scholar] [CrossRef]
- Martino, A.T.; Basner-Tschakarjan, E.; Markusic, D.M.; Finn, J.D.; Hinderer, C.; Zhou, S.; Ostrov, D.A.; Srivastava, A.; Ertl, H.C.; Terhorst, C.; et al. Engineered AAV vector minimizes in vivo targeting of transduced hepatocytes by capsid-specific CD8+ T cells. Blood 2013, 121, 2224–2233. [Google Scholar] [CrossRef]
- Arruda, V.R.; Favaro, P.; Finn, J.D. Strategies to modulate immune responses: A new frontier for gene therapy. Mol. Ther. 2009, 17, 1492–1503. [Google Scholar] [CrossRef]
- Palmer, D.J.; Ng, P. Methods for the production of helper-dependent adenoviral vectors. Methods Mol. Biol. 2008, 433, 33–53. [Google Scholar]
- Koehler, D.R.; Martin, B.; Corey, M.; Palmer, D.; Ng, P.; Tanswell, A.K.; Hu, J. Readministration of helper-dependent adenovirus to mouse lung. Gene Ther. 2006, 13, 773–780. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Schirmbeck, R.; Reimann, J.; Kochanek, S.; Kreppel, F. The immunogenicity of adenovirus vectors limits the multispecificity of CD8 T-cell responses to vector-encoded transgenic antigens. Mol. Ther. 2008, 16, 1609–1616. [Google Scholar] [CrossRef]
- Seregin, S.S.; Appledorn, D.M.; McBride, A.J.; Schuldt, N.J.; Aldhamen, Y.A.; Voss, T.; Wei, J.; Bujold, M.; Nance, W.; Godbehere, S.; et al. Transient pretreatment with glucocorticoid ablates innate toxicity of systemically delivered adenoviral vectors without reducing efficacy. Mol. Ther. 2009, 17, 685–696. [Google Scholar] [CrossRef]
- Cramer, M.L.; Shao, G.; Rodino-Klapac, L.R.; Chicoine, L.G.; Martin, P.T. Induction of T-Cell Infiltration and Programmed Death Ligand 2 Expression by Adeno-Associated Virus in Rhesus Macaque Skeletal Muscle and Modulation by Prednisone. Hum. Gene Ther. 2017, 28, 493–509. [Google Scholar] [CrossRef]
- Majowicz, A.; Salas, D.; Zabaleta, N.; Rodriguez-Garcia, E.; Gonzalez-Aseguinolaza, G.; Petry, H.; Ferreira, V. Successful Repeated Hepatic Gene Delivery in Mice and Non-human Primates Achieved by Sequential Administration of AAV5(ch) and AAV1. Mol. Ther. 2017, 25, 1831–1842. [Google Scholar] [CrossRef]
- Vandenberghe, L.H.; Wilson, J.M.; Gao, G. Tailoring the AAV vector capsid for gene therapy. Gene Ther. 2009, 16, 311–319. [Google Scholar] [CrossRef]
- Bockor, L.; Bortolussi, G.; Iaconcig, A.; Chiaruttini, G.; Tiribelli, C.; Giacca, M.; Benvenuti, F.; Zentilin, L.; Muro, A.F. Repeated AAV-mediated gene transfer by serotype switching enables long-lasting therapeutic levels of hUgt1a1 enzyme in a mouse model of Crigler-Najjar Syndrome Type I. Gene Ther. 2017, 24, 649–660. [Google Scholar] [CrossRef]
- Ahlmann, M.; Hempel, G. The effect of cyclophosphamide on the immune system: Implications for clinical cancer therapy. Cancer Chemother. Pharm. 2016, 78, 661–671. [Google Scholar] [CrossRef]
- Strauss, G.; Osen, W.; Debatin, K.M. Induction of apoptosis and modulation of activation and effector function in T cells by immunosuppressive drugs. Clin. Exp. Immunol. 2002, 128, 255–266. [Google Scholar] [CrossRef]
- Al-Homsi, A.S.; Roy, T.S.; Cole, K.; Feng, Y.; Duffner, U. Post-transplant high-dose cyclophosphamide for the prevention of graft-versus-host disease. Biol. Blood Marrow Transplant. 2015, 21, 604–611. [Google Scholar] [CrossRef] [Green Version]
- Wojcik, R. Reactivity of the immunological system of rats stimulated with Biolex-Beta HP after cyclophosphamide immunosuppression. Cent. Eur. J. Immunol. 2014, 39, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; Reese, T.A. Making Mouse Models That Reflect Human Immune Responses. Trends Immunol. 2017, 38, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, H.; Duan, R.; Hu, J. Overcoming Immunological Challenges to Helper-Dependent Adenoviral Vector-Mediated Long-Term CFTR Expression in Mouse Airways. Genes 2020, 11, 565. https://doi.org/10.3390/genes11050565
Cao H, Duan R, Hu J. Overcoming Immunological Challenges to Helper-Dependent Adenoviral Vector-Mediated Long-Term CFTR Expression in Mouse Airways. Genes. 2020; 11(5):565. https://doi.org/10.3390/genes11050565
Chicago/Turabian StyleCao, Huibi, Rongqi Duan, and Jim Hu. 2020. "Overcoming Immunological Challenges to Helper-Dependent Adenoviral Vector-Mediated Long-Term CFTR Expression in Mouse Airways" Genes 11, no. 5: 565. https://doi.org/10.3390/genes11050565
APA StyleCao, H., Duan, R., & Hu, J. (2020). Overcoming Immunological Challenges to Helper-Dependent Adenoviral Vector-Mediated Long-Term CFTR Expression in Mouse Airways. Genes, 11(5), 565. https://doi.org/10.3390/genes11050565