Molecular Diagnosis and Genetic Counseling of Cystic Fibrosis and Related Disorders: New Challenges


Abstract
:1. Introduction
2. Molecular Diagnosis
2.1. Molecular Diagnosis of CF and CFTR-RD
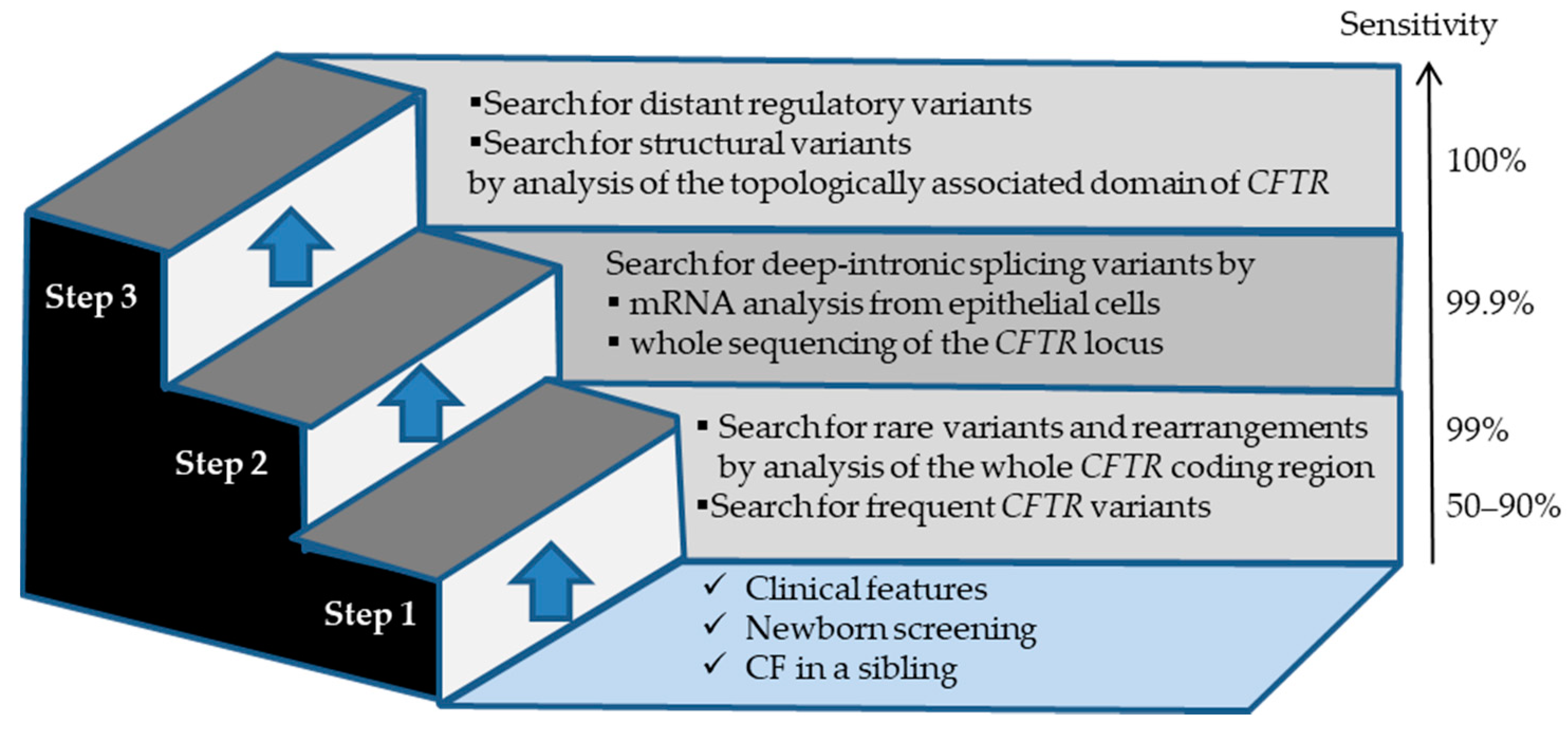
2.2. Tools and Strategies Used for the Molecular Diagnosis of CF and CFTR-RD
2.3. Prenatal and Preimplantation Diagnoses of CF
2.4. Recommendations for Population-Based CF Carrier Screening
2.5. Impact of Variant Heterogeneity on Personalized Medicine
3. How to Assess the Impact of CFTR Variants—The Challenge of Penetrance
3.1. Population Data (Clinical)
3.2. Literature Data
3.3. Computational and Predictive Data
3.4. Allelic and Segregation Data
3.5. Functional Data
3.6. Population Genetics and Penetrance Associated to CFTR Variants
4. The Challenges of Genetic Counseling—When Genetic Counseling Meets Diagnosis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Farrell, P.M.; White, T.B.; Ren, C.L.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of cystic fibrosis: Consensus guidelines from the cystic fibrosis foundation. J. Pediatr. 2017, 181, S4–S15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwiebert, E.M.; Egan, M.E.; Hwang, T.H.; Fulmer, S.B.; Allen, S.S.; Cutting, G.R.; Guggino, W.B. CFTR regulates outwardly rectifying chloride channels through an autocrine mechanism involving ATP. Cell 1995, 81, 1063–1073. [Google Scholar] [CrossRef] [Green Version]
- Bareil, C.; Bergougnoux, A. CFTR gene variants, epidemiology and molecular pathology. Arch. Pédiatrie 2020, 27, eS8–eS12. [Google Scholar] [CrossRef]
- Audrézet, M.P.; Munck, A.; Scotet, V.; Claustres, M.; Roussey, M.; Delmas, D.; Férec, C.; Desgeorges, M. Comprehensive CFTR gene analysis of the French cystic fibrosis screened newborn cohort: Implications for diagnosis, genetic counseling, and mutation-specific therapy. Genet. Med. 2015, 17, 108–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girardet, A.; Viart, V.; Plaza, S.; Daina, G.; De Rycke, M.; Des Georges, M.; Fiorentino, F.; Harton, G.; Ishmukhametova, A.; Navarro, J.; et al. The improvement of the best practice guidelines for preimplantation genetic diagnosis of cystic fibrosis: Toward an international consensus. Eur. J. Hum. Genet. 2016, 24, 469–478. [Google Scholar] [CrossRef]
- Gruber, A.; Pacault, M.; El Khattabi, L.A.; Vaucouleur, N.; Orhant, L.; Bienvenu, T.; Girodon, E.; Vidaud, D.; Leturcq, F.; Costa, C.; et al. Non-invasive prenatal diagnosis of paternally inherited disorders from maternal plasma: Detection of NF1 and CFTR mutations using droplet digital PCR. Clin. Chem. Lab. Med. 2018, 56, 728–738. [Google Scholar] [CrossRef]
- Bombieri, C.; Claustres, M.; De Boeck, K.; Derichs, N.; Dodge, J.; Girodon, E.; Sermet, I.; Schwarz, M.; Tzetis, M.; Wilschanski, M.; et al. Recommendations for the classification of diseases as CFTR-related disorders. J. Cyst. Fibros. 2011, 10, S86–S102. [Google Scholar] [CrossRef] [Green Version]
- Pagin, A.; Sermet-Gaudelus, I.; Burgel, P.-R. Genetic diagnosis in practice: From cystic fibrosis to CFTR-related disorders. Arch. Pédiatrie 2020, 27, eS25–eS29. [Google Scholar] [CrossRef]
- Miller, A.C.; Comellas, A.P.; Hornick, D.B.; Stoltz, D.A.; Cavanaugh, J.E.; Gerke, A.K.; Welsh, M.J.; Zabner, J.; Polgreen, P.M. Cystic fibrosis carriers are at increased risk for a wide range of cystic fibrosis-related conditions. Proc. Natl. Acad. Sci. USA 2020, 117, 1621–1627. [Google Scholar] [CrossRef] [Green Version]
- Pagin, A.; Bergougnoux, A.; Girodon, E.; Reboul, M.; Willoquaux, C.; Kesteloot, M.; Raynal, C.; Bienvenu, T.; Humbert, M.; Lalau, G.; et al. Novel ADGRG2 truncating variants in patients with X-linked congenital absence of vas deferens. Andrology 2020, 8, 618–624. [Google Scholar] [CrossRef]
- Masson, E.; Chen, J.-M.; Audrézet, M.-P.; Cooper, D.N.; Férec, C. A conservative assessment of the major genetic causes of idiopathic chronic pancreatitis: Data from a comprehensive analysis of PRSS1, SPINK1, CTRC and CFTR genes in 253 young French patients. PLoS ONE 2013, 8, e73522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellani, C.; Cuppens, H.; Macek, M.; Cassiman, J.J.; Kerem, E.; Durie, P.; Tullis, E.; Assael, B.M.; Bombieri, C.; Brown, A.; et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J. Cyst. Fibros. 2008, 7, 179–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dequeker, E.; Stuhrmann, M.; Morris, M.A.; Casals, T.; Castellani, C.; Claustres, M.; Cuppens, H.; des Georges, M.; Ferec, C.; Macek, M.; et al. Best practice guidelines for molecular genetic diagnosis of cystic fibrosis and CFTR-related disorders—Updated European recommendations. Eur. J. Hum. Genet. 2009, 17, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Ratbi, I.; Legendre, M.; Niel, F.; Martin, J.; Soufir, J.-C.; Izard, V.; Costes, B.; Costa, C.; Goossens, M.; Girodon, E. Detection of cystic fibrosis transmembrane conductance regulator (CFTR) gene rearrangements enriches the mutation spectrum in congenital bilateral absence of the vas deferens and impacts on genetic counselling. Hum. Reprod. 2007, 22, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Raynal, C.; Girodon, E.; Audrezet, M.P.; Cabet, F.; Pagin, A.; Reboul, M.P.; Dufernez, F.; Fergelot, P.; Bergougnoux, A.; Fanen, P.; et al. CFTR gene variants: A predisposition factor to aquagenic palmoplantar keratoderma. Br. J. Dermatol. 2019, 181, 1097–1099. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Mutation Data Base (CFMDB). Available online: http://www.genet.sickkids.on.ca/ (accessed on 7 May 2020).
- The Molecular Genetic Epidemiology of Cystic Fibrosis: Report of a Joint Meeting of WHO/ECFTN/ICF(M)A/ECFS, Genoa, Italy, 19 June 2002. Available online: https://apps.who.int/iris/handle/10665/68702 (accessed on 7 May 2020).
- Kılınç, M.O.; Ninis, V.N.; Dağlı, E.; Demirkol, M.; Özkınay, F.; Arıkan, Z.; Çoğulu, Ö.; Hüner, G.; Karakoç, F.; Tolun, A. Highest heterogeneity for cystic fibrosis: 36 mutations account for 75% of all CF chromosomes in Turkish patients. Am. J. Med. Genet. 2002, 113, 250–257. [Google Scholar] [CrossRef]
- Bergougnoux, A.; Taulan-Cadars, M.; Claustres, M.; Raynal, C. Current and future molecular approaches in the diagnosis of cystic fibrosis. Expert Rev. Respir. Med. 2018, 12, 415–426. [Google Scholar] [CrossRef]
- Kharrazi, M.; Yang, J.; Bishop, T.; Lessing, S.; Young, S.; Graham, S.; Pearl, M.; Chow, H.; Ho, T.; Currier, R.; et al. Newborn screening for cystic fibrosis in California. Pediatrics 2015, 136, 1062–1072. [Google Scholar] [CrossRef] [Green Version]
- Faà, V.; Incani, F.; Meloni, A.; Corda, D.; Masala, M.; Baffico, A.M.; Seia, M.; Cao, A.; Rosatelli, M.C. Characterization of a disease-associated mutation affecting a putative splicing regulatory element in intron 6b of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Gene. J. Biol. Chem. 2009, 284, 30024–30031. [Google Scholar] [CrossRef] [Green Version]
- Costa, C.; Pruliere-Escabasse, V.; de Becdelievre, A.; Gameiro, C.; Golmard, L.; Guittard, C.; Bassinet, L.; Bienvenu, T.; Georges, M.D.; Epaud, R.; et al. A recurrent deep-intronic splicing CF mutation emphasizes the importance of mRNA studies in clinical practice. J. Cyst. Fibros. 2011, 10, 479–482. [Google Scholar] [CrossRef] [Green Version]
- Bonini, J.; Varilh, J.; Raynal, C.; Thèze, C.; Beyne, E.; Audrezet, M.-P.; Ferec, C.; Bienvenu, T.; Girodon, E.; Tuffery-Giraud, S.; et al. Small-scale high-throughput sequencing–based identification of new therapeutic tools in cystic fibrosis. Genet. Med. 2015, 17, 796–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergougnoux, A.; Délétang, K.; Pommier, A.; Varilh, J.; Houriez, F.; Altieri, J.P.; Koenig, M.; Férec, C.; Claustres, M.; Lalau, G.; et al. Functional characterization and phenotypic spectrum of three recurrent disease-causing deep intronic variants of the CFTR gene. J. Cyst. Fibros. 2019, 18, 468–475. [Google Scholar] [CrossRef]
- Moisan, S.; Berlivet, S.; Ka, C.; Gac, G.L.; Dostie, J.; Férec, C. Analysis of long-range interactions in primary human cells identifies cooperative CFTR regulatory elements. Nucleic Acids Res. 2016, 44, 2564–2576. [Google Scholar] [CrossRef] [Green Version]
- Guissart, C.; Dubucs, C.; Raynal, C.; Girardet, A.; Tran Mau Them, F.; Debant, V.; Rouzier, C.; Boureau-Wirth, A.; Haquet, E.; Puechberty, J.; et al. Non-invasive prenatal diagnosis (NIPD) of cystic fibrosis: An optimized protocol using MEMO fluorescent PCR to detect the p.Phe508del mutation. J. Cyst. Fibros. 2017, 16, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Guissart, C.; Them, F.T.M.; Debant, V.; Viart, V.; Dubucs, C.; Pritchard, V.; Rouzier, C.; Boureau-Wirth, A.; Haquet, E.; Puechberty, J.; et al. A broad test based on fluorescent-multiplex PCR for noninvasive prenatal diagnosis of Cystic Fibrosis. Fetal Diagn. Ther. 2019, 45, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Bergougnoux, A.; Jouannic, J.-M.; Verneau, F.; Bienvenu, T.; Gaitch, N.; Raynal, C.; Girodon, E. Isolated nonvisualization of the fetal gallbladder should be considered for the prenatal diagnosis of Cystic Fibrosis. Fetal Diagn. Ther. 2019, 45, 312–316. [Google Scholar] [CrossRef]
- Richards, C.S.; Bradley, L.A.; Amos, J.; Allitto, B.; Grody, W.W.; Maddalena, A.; McGinnis, M.J.; Prior, T.W.; Popovich, B.W.; Watson, M.S. Standards and Guidelines for CFTR Mutation Testing. Genet. Med. 2002, 4, 379–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, M.S.; Cutting, G.R.; Desnick, R.J.; Driscoll, D.A.; Klinger, K.; Mennuti, M.; Palomaki, G.E.; Popovich, B.W.; Pratt, V.M.; Rohlfs, E.M.; et al. Cystic fibrosis population carrier screening: 2004 revision of American college of medical genetics mutation panel. Genet. Med. 2004, 6, 387–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, C.; Pepper, M.S. Cystic fibrosis on the African continent. Genet. Med. 2016, 18, 653–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammoudeh, S.; Gadelhak, W.; AbdulWahab, A.; Al-Langawi, M.; Janahi, I.A. Approaching two decades of cystic fibrosis research in Qatar: A historical perspective and future directions. Multidiscip. Respir. Med. 2019, 14, 29. [Google Scholar] [CrossRef]
- Welsh, M.J.; Smith, A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993, 73, 1251–1254. [Google Scholar] [CrossRef]
- Highsmith, W.E.; Burch, L.H.; Zhou, Z.; Olsen, J.C.; Boat, T.E.; Spock, A.; Gorvoy, J.D.; Quittell, L.; Friedman, K.J.; Silverman, L.M.; et al. A novel mutation in the cystic fibrosis gene in patients with pulmonary disease but normal sweat chloride concentrations. N. Engl. J. Med. 1994, 331, 974–980. [Google Scholar] [CrossRef]
- Haardt, M.; Benharouga, M.; Lechardeur, D.; Kartner, N.; Lukacs, G.L. C-terminal truncations destabilize the Cystic Fibrosis Transmembrane Conductance Regulator without impairing its biogenesis. A novel class of mutation. J. Biol. Chem. 1999, 274, 21873–21877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Sadeq, D.; Abunada, T.; Dalloul, R.; Fahad, S.; Taleb, S.; Aljassim, K.; Al Hamed, F.A.; Zayed, H. Spectrum of mutations of cystic fibrosis in the 22 Arab countries: A systematic review. Respirology 2019, 24, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Fajac, I.; Girodon, E. Genomically-guided therapies: A new era for cystic fibrosis. Arch. Pédiatrie 2020, 27, eS41–eS44. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Sosnay, P.R.; Siklosi, K.R.; Van Goor, F.; Kaniecki, K.; Yu, H.; Sharma, N.; Ramalho, A.S.; Amaral, M.D.; Dorfman, R.; Zielenski, J.; et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat. Genet. 2013, 45, 1160–1167, CFTR2. Available online: https://www.cftr2.org/ (accessed on 7 May 2020). [CrossRef] [Green Version]
- Claustres, M.; Thèze, C.; des Georges, M.; Baux, D.; Girodon, E.; Bienvenu, T.; Audrezet, M.-P.; Dugueperoux, I.; Férec, C.; Lalau, G.; et al. CFTR-France, a national relational patient database for sharing genetic and phenotypic data associated with rare CFTR variants. Hum. Mutat. 2017, 38, 1297–1315. Available online: https://cftr.iurc.montp.inserm.fr/cgi-bin/home.cgi (accessed on 7 May 2020). [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 7 May 2020). [CrossRef] [Green Version]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. Available online: http://www.hgmd.cf.ac.uk/ac/index.php (accessed on 7 May 2020). [CrossRef] [PubMed] [Green Version]
- Boussaroque, A.; Bergougnoux, A.; Raynal, C.; Audrézet, M.-P.; Sasorith, S.; Férec, C.; Bienvenu, T.; Girodon, E. Pitfalls in the interpretation of CFTR variants in the context of incidental findings. Hum. Mutat. 2019, 40, 2239–2246. [Google Scholar] [CrossRef]
- Leiden Open Version Database (LOVD). Available online: https://www.lovd.nl/ (accessed on 7 May 2020).
- Sasorith, S.; Baux, D.; Bergougnoux, A.; Paulet, D.; Lahure, A.; Bareil, C.; Taulan-Cadars, M.; Roux, A.; Koenig, M.; Claustres, M.; et al. The CYSMA web server: An example of integrative tool for in silico analysis of missense variants identified in Mendelian disorders. Hum. Mutat. 2020, 41, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. Available online: http://varsome.com/ (accessed on 7 May 2020). [CrossRef] [PubMed]
- Li, Q.; Wang, K. InterVar: Clinical interpretation of genetic variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. Available online: http://wintervar.wglab.org/ (accessed on 7 May 2020). [CrossRef] [PubMed] [Green Version]
- Vecchio-Pagán, B.; Blackman, S.M.; Lee, M.; Atalar, M.; Pellicore, M.J.; Pace, R.G.; Franca, A.L.; Raraigh, K.S.; Sharma, N.; Knowles, M.R.; et al. Deep resequencing of CFTR in 762 F508del homozygotes reveals clusters of non-coding variants associated with cystic fibrosis disease traits. Hum. Genome Var. 2016, 3, 16038. [Google Scholar] [CrossRef] [Green Version]
- Bergougnoux, A.; Boureau-Wirth, A.; Rouzier, C.; Altieri, J.-P.; Verneau, F.; Larrieu, L.; Koenig, M.; Claustres, M.; Raynal, C. A false positive newborn screening result due to a complex allele carrying two frequent CF-causing variants. J. Cyst. Fibros. 2016, 15, 309–312. [Google Scholar] [CrossRef]
- El-Seedy, A.; Girodon, E.; Norez, C.; Pajaud, J.; Pasquet, M.-C.; de Becdelièvre, A.; Bienvenu, T.; des Georges, M.; Cabet, F.; Lalau, G.; et al. CFTR mutation combinations producing frequent complex alleles with different clinical and functional outcomes. Hum. Mutat. 2012, 33, 1557–1565. [Google Scholar] [CrossRef]
- Chevalier, B.; Hinzpeter, A. The influence of CFTR complex alleles on precision therapy of cystic fibrosis. J. Cyst. Fibros. 2020, 19, S15–S18. [Google Scholar] [CrossRef] [Green Version]
- Křenková, P.; Piskáčková, T.; Holubová, A.; Balaščaková, M.; Krulišová, V.; Čamajová, J.; Turnovec, M.; Libik, M.; Norambuena, P.; Štambergová, A.; et al. Distribution of CFTR mutations in the Czech population: Positive impact of integrated clinical and laboratory expertise, detection of novel/de novo alleles and relevance for related/derived populations. J. Cyst. Fibros. 2013, 12, 532–537. [Google Scholar] [CrossRef] [Green Version]
- Niel, F.; Legendre, M.; Bienvenu, T.; Bieth, E.; Lalau, G.; Sermet, I.; Bondeux, D.; Boukari, R.; Derelle, J.; Levy, P.; et al. A new large CFTR rearrangement illustrates the importance of searching for complex alleles. Hum. Mutat. 2006, 27, 716–717. [Google Scholar] [CrossRef] [PubMed]
- Quinton, P.; Molyneux, L.; Ip, W.; Dupuis, A.; Avolio, J.; Tullis, E.; Conrad, D.; Shamsuddin, A.K.; Durie, P.; Gonska, T. β-Adrenergic sweat secretion as a diagnostic test for Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; Wiegerinck, C.L.; de Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; de Winter-de Groot, K.M.; Brandsma, A.M.; de Jong, N.W.M.; Bijvelds, M.J.C.; Scholte, B.J.; et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Awatade, N.T.; Wong, S.L.; Hewson, C.K.; Fawcett, L.K.; Kicic, A.; Jaffe, A.; Waters, S.A. Human primary epithelial cell models: Promising tools in the era of cystic fibrosis personalized medicine. Front. Pharmacol. 2018, 9, 1429. [Google Scholar] [CrossRef] [Green Version]
- Pranke, I.M.; Hatton, A.; Simonin, J.; Jais, J.P.; Le Pimpec-Barthes, F.; Carsin, A.; Bonnette, P.; Fayon, M.; Stremler-Le Bel, N.; Grenet, D.; et al. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Sci. Rep. 2017, 7, 7375. [Google Scholar] [CrossRef]
- Amaral, M.D.; de Boeck, K.; Amaral, M.; Davies, J.C.; de Boeck, K.; Drevinek, P.; Elborn, S.; Kerem, E.; Lee, T. Theranostics by testing CFTR modulators in patient-derived materials: The current status and a proposal for subjects with rare CFTR mutations. J. Cyst. Fibros. 2019, 18, 685–692. [Google Scholar] [CrossRef]
- Raynal, C.; Baux, D.; Theze, C.; Bareil, C.; Taulan, M.; Roux, A.-F.; Claustres, M.; Tuffery-Giraud, S.; des Georges, M. A classification model relative to splicing for variants of unknown clinical significance: Application to the CFTR gene. Hum. Mutat. 2013, 34, 774–784. [Google Scholar] [CrossRef]
- Sharma, N.; Sosnay, P.R.; Ramalho, A.S.; Douville, C.; Franca, A.; Gottschalk, L.B.; Park, J.; Lee, M.; Vecchio-Pagan, B.; Raraigh, K.S.; et al. Experimental assessment of splicing variants using expression Minigenes and comparison with in Silico predictions. Hum. Mutat. 2014, 35, 1249–1259. [Google Scholar] [CrossRef]
- Amato, F.; Scudieri, P.; Musante, I.; Tomati, V.; Caci, E.; Comegna, M.; Maietta, S.; Manzoni, F.; Di Lullo, A.M.; Wachter, E.; et al. Two CFTR mutations within codon 970 differently impact on the chloride channel functionality. Hum. Mutat. 2019, 40, 742–748. [Google Scholar] [CrossRef]
- Ramalho, A.S.; Clarke, L.A.; Sousa, M.; Felicio, V.; Barreto, C.; Lopes, C.; Amaral, M.D. Comparative ex vivo, in vitro and in silico analyses of a CFTR splicing mutation: Importance of functional studies to establish disease liability of mutations. J. Cyst. Fibros. 2016, 15, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Hinzpeter, A.; Aissat, A.; Sondo, E.; Costa, C.; Arous, N.; Gameiro, C.; Martin, N.; Tarze, A.; Weiss, L.; de Becdelièvre, A.; et al. Alternative splicing at a NAGNAG acceptor site as a novel phenotype modifier. PLoS Genet. 2010, 6, e1001153. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Groman, J.D.; Hefferon, T.W.; Casals, T.; Bassas, L.; Estivill, X.; Des Georges, M.; Guittard, C.; Koudova, M.; Fallin, M.D.; Nemeth, K.; et al. Variation in a repeat sequence determines whether a common variant of the cystic fibrosis transmembrane conductance regulator gene is pathogenic or benign. Am. J. Hum. Genet. 2004, 74, 176–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thauvin-Robinet, C.; Munck, A.; Huet, F.; Génin, E.; Bellis, G.; Gautier, E.; Audrézet, M.-P.; Férec, C.; Lalau, G.; Des Georges, M.; et al. The very low penetrance of cystic fibrosis for the R117H mutation: A reappraisal for genetic counselling and newborn screening. J. Med. Genet. 2009, 46, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Boussaroque, A.; Audrézet, M.-P.; Raynal, C.; Sermet-Gaudelus, I.; Bienvenu, T.; Férec, C.; Bergougnoux, A.; Lopez, M.; Scotet, V.; Munck, A.; et al. Penetrance is a critical parameter for assessing the disease liability of CFTR variants. J. Cyst. Fibros. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bieth, E.; Nectoux, J.; Girardet, A.; Gruchy, N.; Mittre, H.; Laurans, M.; Guenet, D.; Brouard, J.; Gerard, M. Genetic counseling for cystic fibrosis: A basic model with new challenges. Arch. Pédiatrie 2020, 27, eS30–eS34. [Google Scholar] [CrossRef]
- Nijmeijer, S.C.M.; Conijn, T.; Lakeman, P.; Henneman, L.; Wijburg, F.A.; Haverman, L. Attitudes of the general population towards preconception expanded carrier screening for autosomal recessive disorders including inborn errors of metabolism. Mol. Genet. Metab. 2019, 126, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Van Steijvoort, E.; Chokoshvili, D.; Cannon, J.W.; Peeters, H.; Peeraer, K.; Matthijs, G.; Borry, P. Interest in expanded carrier screening among individuals and couples in the general population: Systematic review of the literature. Hum. Reprod. Update 2020, 26, 335–355. [Google Scholar] [CrossRef]
- Delatycki, M.B.; Alkuraya, F.; Archibald, A.; Castellani, C.; Cornel, M.; Grody, W.W.; Henneman, L.; Ioannides, A.S.; Kirk, E.; Laing, N.; et al. International perspectives on the implementation of reproductive carrier screening. Prenat. Diagn. 2020, 40, 301–310. [Google Scholar] [CrossRef]
- Bergougnoux, A.; Lopez, M.; Girodon, E. The role of extended CFTR gene sequencing in newborn screening for Cystic Fibrosis. Int. J. Neonatal Screen. 2020, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Farrell, P.M.; Rock, M.J.; Baker, M.W. The impact of the CFTR gene discovery on Cystic Fibrosis diagnosis, counseling, and preventive therapy. Genes 2020, 11, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceyhan-Birsoy, O.; Murry, J.B.; Machini, K.; Lebo, M.S.; Yu, T.W.; Fayer, S.; Genetti, C.A.; Schwartz, T.S.; Agrawal, P.B.; Parad, R.B.; et al. Interpretation of genomic sequencing results in healthy and ill newborns: Results from the BabySeq Project. Am. J. Hum. Genet. 2019, 104, 76–93. [Google Scholar] [CrossRef] [Green Version]
- Kingsmore, S.F. Newborn testing and screening by whole-genome sequencing. Genet. Med. 2016, 18, 214–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcken, B.; Wiley, V. Fifty years of newborn screening. J. Paediatr. Child Health 2015, 51, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Cazacu, I.M.; Farkas, N.; Garami, A.; Balaskó, M.; Mosdósi, B.; Alizadeh, H.; Gyöngyi, Z.; Rakonczay, Z.; Vigh, É.; Habon, T.; et al. Pancreatitis-Associated Genes and pancreatic cancer risk: A systematic review and meta-analysis. Pancreas 2018, 47, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.; Férec, C.; Macek, M.; Frischer, T.; Renner, S.; Riss, K.; Barton, D.; Repetto, T.; Tzetis, M.; Giteau, K.; et al. Estimating the age of p.(Phe508del) with family studies of geographically distinct European populations and the early spread of cystic fibrosis. Eur. J. Hum. Genet. 2018, 26, 1832–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Categories of Evidence | CFTR-France | CFTR2 | ||
|---|---|---|---|---|
| Population data: general population | + | Link to dbSNP and gnomAD | + | Reference to general population and CF carriers analysis for incomplete penetrance of CFTR variants |
| Population data: clinical observations | + | - 853 variants in about 5000 CF and CFTR-RD patients, and asymptomatic compound heterozygous individuals (data collected in molecular genetics laboratories, cross-reference with the French CF Registry) - Per patient: Age at diagnosis, symptoms, pancreatic status, meconium ileus, sweat chloride values, NBS - Link to CF Mutation Database and CFTR2 | + | - 432 variants in about 89,000 CF patients (data collected from national registries) - Aggregated data for a given variant or genotype: age, lung and pancreatic function, Pseudomonas aeruginosa infection, sweat chloride values - Reference to ClinVar and LOVD |
| Literature | + | Link to PubMed for functional data | + | Link to PubMed for clinical and functional data |
| Computational predictions | + | AGVGD, MAPP, SIFT, PolyPhen-2, CYSMA | - | |
| Allelic and segregation data | + | - Data on variants identified in trans - Data on complex alleles | + | - Data provided on specific genotypes - No data on complex alleles |
| Functional data | + | Link to PubMed (transcript and protein studies) | + | - Data on CFTR protein maturation, folding, quantity and function in different cell lines - Link to PubMed |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bienvenu, T.; Lopez, M.; Girodon, E. Molecular Diagnosis and Genetic Counseling of Cystic Fibrosis and Related Disorders: New Challenges. Genes 2020, 11, 619. https://doi.org/10.3390/genes11060619
Bienvenu T, Lopez M, Girodon E. Molecular Diagnosis and Genetic Counseling of Cystic Fibrosis and Related Disorders: New Challenges. Genes. 2020; 11(6):619. https://doi.org/10.3390/genes11060619
Chicago/Turabian StyleBienvenu, Thierry, Maureen Lopez, and Emmanuelle Girodon. 2020. "Molecular Diagnosis and Genetic Counseling of Cystic Fibrosis and Related Disorders: New Challenges" Genes 11, no. 6: 619. https://doi.org/10.3390/genes11060619
APA StyleBienvenu, T., Lopez, M., & Girodon, E. (2020). Molecular Diagnosis and Genetic Counseling of Cystic Fibrosis and Related Disorders: New Challenges. Genes, 11(6), 619. https://doi.org/10.3390/genes11060619