Rapid Direct Nucleic Acid Amplification Test without RNA Extraction for SARS-CoV-2 Using a Portable PCR Thermocycler


Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and Controls Used for Assay Development
2.2. Collection and Processing of Sputum and Nasal Exudate Samples
2.3. Primer and Probes
2.4. RT-qPCR and DIRECT-PCR
2.5. DIRECT-PCR of SARS-CoV-2 N Gene in Sputum and Nasal Exudate
2.6. Optimization of Fast DIRECT-PCR Assay
2.7. Performance of Portable Real-Time Thermocycler
2.8. Statistical Analysis
3. Results
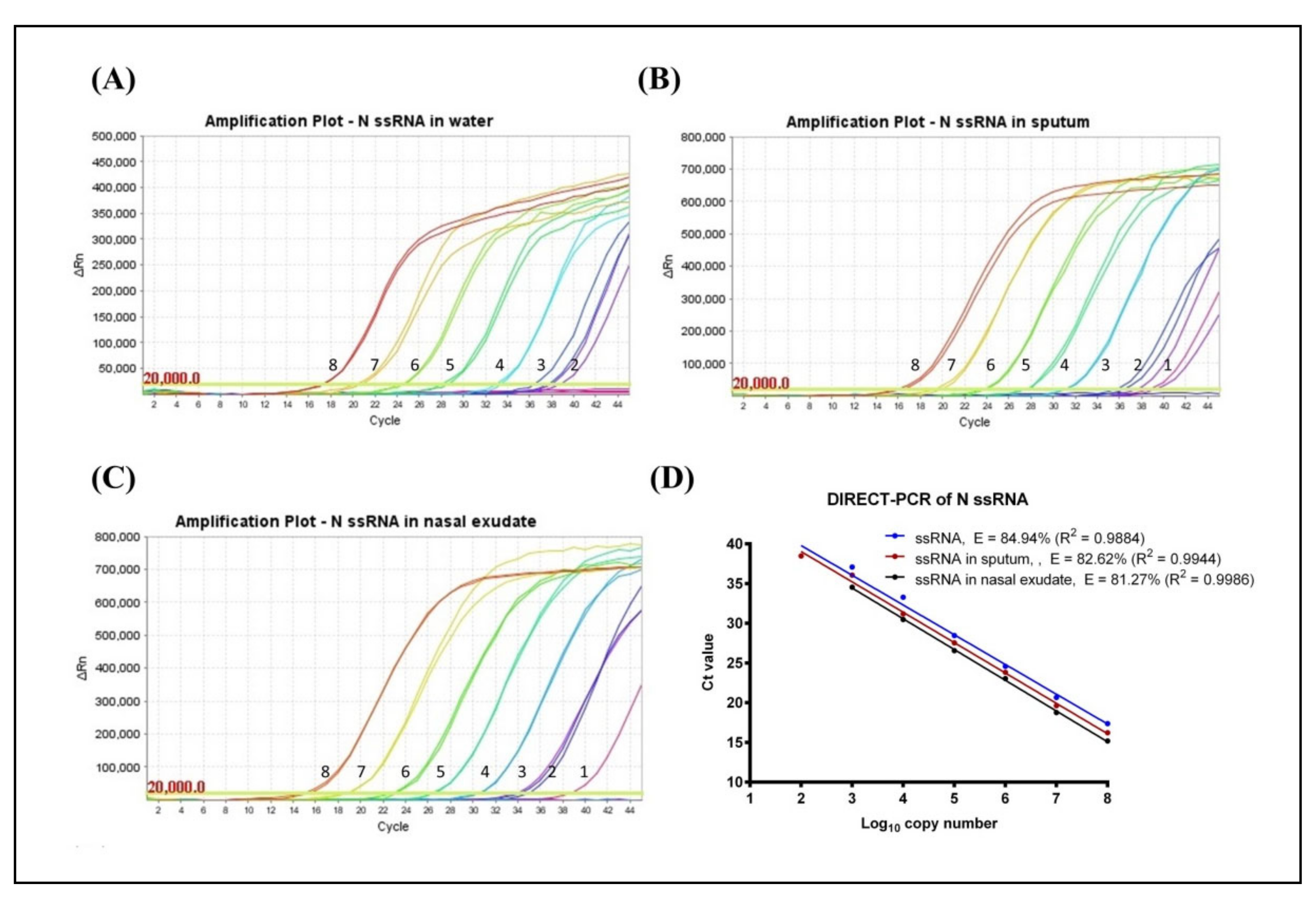
3.1. Determination of LoD and Amplification Efficiency (E)
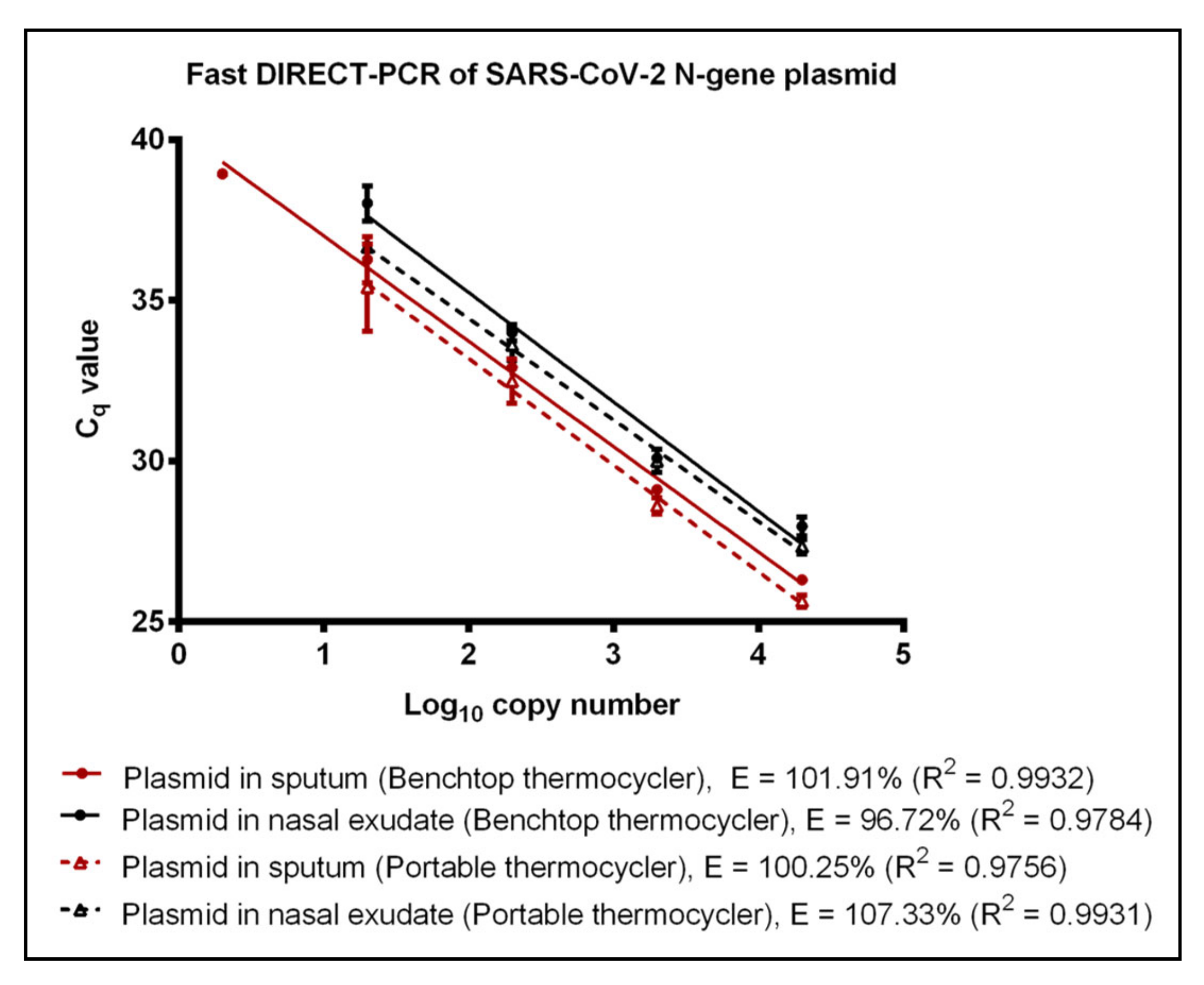
3.2. Evaluation of Fast DIRECT-PCR Assay
3.3. Performance of Portable Thermocycler
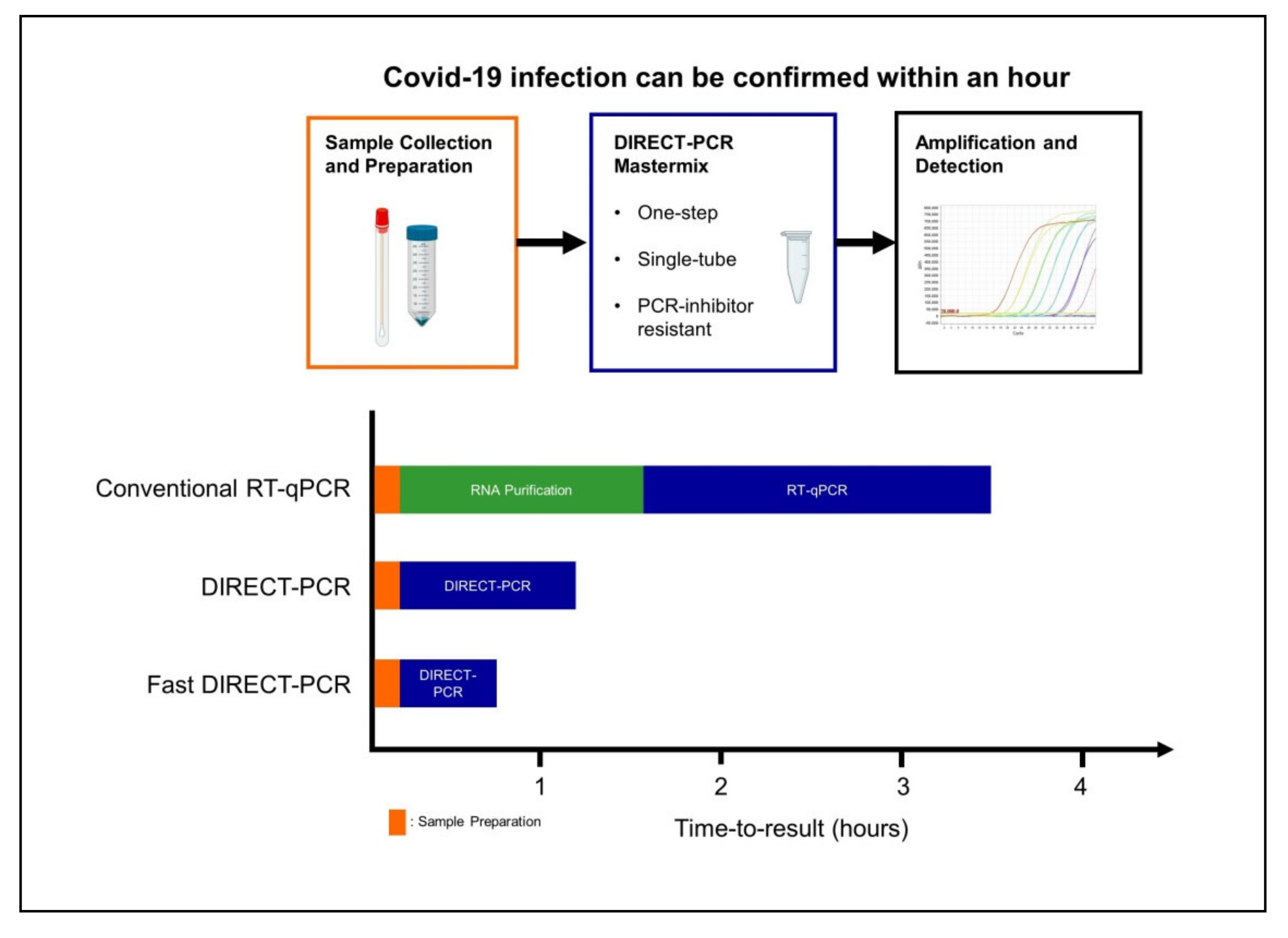
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. New Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Director-General’s opening remarks at the media briefing on COVID-19 - 11 March 2020. Available online: https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (accessed on 12 March 2020).
- WHO. COVID-19 Dashboard. Available online: https://covid19.who.int/ (accessed on 16 April 2020).
- Beeching, N.J.; E Fletcher, T.; Beadsworth, M.B.J. Covid-19: Testing times. BMJ 2020, 369, m1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, Y.; Li, Z.; Chua, Y.X.; Chaw, W.L.; Zhao, Z.; Er, B.; Pung, R.; Chiew, C.J.; Lye, D.C.; Heng, D.; et al. Evaluation of the Effectiveness of Surveillance and Containment Measures for the First 100 Patients with COVID-19 in Singapore — January 2–February 29, 2020. MMWR. Morb. Mortal. Wkly. Rep. 2020, 69, 307–311. [Google Scholar] [CrossRef]
- Wong, J.E.L.; Leo, Y.S.; Tan, C.C. COVID-19 in Singapore—Current Experience. JAMA 2020, 323, 1243. [Google Scholar] [CrossRef]
- MOH. Updates on COVID-19 (Coronavirus Disease 2019) Local Situation. Available online: https://www.moh.gov.sg/covid-19 (accessed on 16 April 2020).
- Zhang, Y.-Z. Novel 2019 coronavirus genome. Available online: http://virological.org/t/novel-2019-coronavirus-genome/319 (accessed on 22 March 2020).
- WHO. Coronavirus disease (COVID-19) technical guidance: Laboratory testing for 2019-nCoV in humans. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance/laboratory-guidance (accessed on 26 March 2020).
- NUS. SSHSPH COVID-19 Science Report; Saw Swee Hock School of Public Health, National University of Singapore: Singapore, 2020. [Google Scholar]
- WHO. Laboratory testing for 2019 novel coronavirus (2019-nCoV) in suspected human cases; WHO/COVID-19/laboratory/2020.5; World Health Organization: 2020. Available online: https://www.who.int/publications/i/item/10665-331501 (accessed on 20 April 2020).
- Nguyen, T.; Bang, D.D.; Wolff, A. 2019 Novel Coronavirus Disease (COVID-19): Paving the Road for Rapid Detection and Point-of-Care Diagnostics. Micromachines 2020, 11, 306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.E.; Li, Z.; Chiew, C.J.; Yong, S.E.; Toh, M.P.; Lee, V.J. Presymptomatic Transmission of SARS-CoV-2 — Singapore, January 23–March 16, 2020. MMWR. Morb. Mortal. Wkly. Rep. 2020, 69, 411–415. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Lau, E.H.; Wu, P.; Deng, X.; Wang, J.; Hao, X.; Lau, Y.C.; Wong, J.Y.; Guan, Y.; Tan, X.; et al. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat. Med. 2020, 26, 672–675. [Google Scholar] [CrossRef] [Green Version]
- Gudbjartsson, D.F.; Helgason, A.; Jonsson, H.; Magnusson, O.T.; Melsted, P.; Norddahl, G.L.; Saemundsdottir, J.; Sigurdsson, A.; Sulem, P.; Agustsdottir, A.B.; et al. Spread of SARS-CoV-2 in the Icelandic Population. New Engl. J. Med. 2020, 382, 2302–2315. [Google Scholar] [CrossRef]
- Sheridan, C. Fast, portable tests come online to curb coronavirus pandemic. Nat. Biotechnol. 2020, 38, 515–518. [Google Scholar] [CrossRef]
- Patel, R.; Babady, N.E.; Theel, E.S.; Storch, G.A.; Pinsky, B.A.; George, K.S.; Smith, T.C.; Bertuzzi, S. Report from the American Society for Microbiology COVID-19 International Summit, 23 March 2020: Value of Diagnostic Testing for SARS–CoV-2/COVID-19. mBio. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Sivalingam, S.P.; Ayi, T.C.; Mustaffa, S.B.; Loo, S.; Tan, B.H.; Yap, E.P.H. Direct RT-PCR detection of Dengue 1-4 virus from crude samples for rapid point-of-care diagnosis. In Proceedings of the SingHealth Duke-NUS Scientific Congress, Singapore, 23–24 September 2016. [Google Scholar]
- Mehta, N.; Perrais, B.; Martin, K.; Kumar, A.; Hobman, T.C.; Cabalfin-Chua, M.N.; Donaldo, M.E.; Painaga, M.S.S.; Gaite, J.Y.; Tran, V.; et al. A Direct from Blood/Plasma Reverse Transcription–Polymerase Chain Reaction for Dengue Virus Detection in Point-of-Care Settings. Am. J. Trop. Med. Hyg. 2019, 100, 1534–1540. [Google Scholar] [CrossRef]
- Pastorino, B.; Bessaud, M.; Grandadam, M.; Murri, S.; Tolou, H.J.; Peyrefitte, C.N. Development of a TaqMan® RT-PCR assay without RNA extraction step for the detection and quantification of African Chikungunya viruses. J. Virol. Methods 2005, 124, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, N.; Nakayama, H.; Yoshizumi, S.; Miyoshi, M.; Tonoike, H.; Shirasaki, Y.; Kojima, K.; Ishida, S. Detection of noroviruses in fecal specimens by direct RT-PCR without RNA purification. J. Virol. Methods 2010, 163, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Bachofen, C.; Willoughby, K.; Zadoks, R.; Burr, P.; Mellor, D.; Russell, G.C. Direct RT-PCR from serum enables fast and cost-effective phylogenetic analysis of bovine viral diarrhoea virus. J. Virol. Methods 2013, 190, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; He, J.-A.; Wang, W.; Xia, Y.; Song, L.; Chen, Z.-H.; Zuo, H.-Z.; Tan, X.-P.; Ho, A.H.-P.; Kong, S.-K.; et al. Development of a direct reverse-transcription quantitative PCR (dirRT-qPCR) assay for clinical Zika diagnosis. Int. J. Infect. Dis. 2019, 85, 167–174. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.T.; Zovanyi, A.M.; Christensen, D.R.; Koehler, J.W.; Minogue, T.D. Evaluation of Inhibitor-Resistant Real-Time PCR Methods for Diagnostics in Clinical and Environmental Samples. PLoS ONE 2013, 8, e73845. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus–Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061. [Google Scholar] [CrossRef]
- CDC, C. Specific primers and probes for detection 2019 novel coronavirus. Available online: http://ivdc.chinacdc.cn/kyjz/202001/t20200121_211337.html (accessed on 1 February 2020).
- CDC. Research Use Only 2019-Novel Coronavirus (2019-nCoV) Real-time RT-PCR Primer and Probe Information. Available online: https://www.cdc.gov/coronavirus/2019-ncov/lab/rt-pcr-panel-primer-probes.html (accessed on 2 March 2020).
- Yang, Y.; Yang, M.; Shen, C.; Wang, F.; Jing, Y.; Li, J.-X.; Zhang, M.; Wang, Z.; Xing, L.; Wei, J.; et al. Evaluating the accuracy of different respiratory specimens in the laboratory diagnosis and monitoring the viral shedding of 2019-nCoV infections 2020. MedRxiv 2020. [Google Scholar]
- Pan, Y.; Zhang, D.; Yang, P.; Poon, L.L.; Wang, Q. Viral load of SARS-CoV-2 in clinical samples. Lancet Infect. Dis. 2020, 20, 411–412. [Google Scholar] [CrossRef]
- Zou, L.; Ruan, F.; Huang, M.; Liang, L.; Huang, H.; Hong, Z.; Yu, J.; Kang, M.; Song, Y.; Xia, J.; et al. SARS-CoV-2 Viral Load in Upper Respiratory Specimens of Infected Patients. New Engl. J. Med. 2020, 382, 1177–1179. [Google Scholar] [CrossRef]
- WHO. COVID 19 Public Health Emergency of International Concern (PHEIC) - Global research and innovation forum: Towards a research roadmap. Available online: https://www.who.int/blueprint/priority-diseases/key-action/Global_Research_Forum_FINAL_VERSION_for_web_14_feb_2020.pdf?ua=1 (accessed on 14 April 2020).
- Bruce, E.A.; Huang, M.-L.; Perchetti, G.A.; Tighe, S.W.; Laaguiby, P.; Hoffman, J.J.; Gerrard, D.L.; Nalla, A.K.; Wei, Y.; Greninger, A.L.; et al. DIRECT RT-qPCR DETECTION OF SARS-CoV-2 RNA FROM PATIENT NASOPHARYNGEAL SWABS WITHOUT AN RNA EXTRACTION STEP 2020. BioRxiv 2020. [Google Scholar]
- Sentmanat, M.; Kouranova, E.; Cui, X. One-step RNA extraction for RT-qPCR detection of 2019-nCoV 2020. BioRxiv 2020. [Google Scholar]
- Arumugam, A.; Wong, S.S. The Potential Use of Unprocessed Sample for RT-qPCR Detection of COVID-19 without an RNA Extraction Step 2020. BioRxiv 2020. [Google Scholar]
- Grant, P.R.; Turner, M.; Shin, G.Y.; Nastouli, E.; Levett, L. Extraction-free COVID-19 (SARS-CoV-2) diagnosis by RT-PCR to increase capacity for national testing programmes during a pandemic. In bioRxiv 2020; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2020. [Google Scholar]
- Merindol, N.; Pépin, G.; Marchand, C.; Rheault, M.; Peterson, C.; Poirier, A.; Germain, H.; Danylo, A. Optimization of SARS-CoV-2 detection by RT-QPCR without RNA extraction 2020. bioRxiv 2020. [Google Scholar]
- Smyrlaki, I.; Ekman, M.; Lentini, A.; Vondracek, M.; Papanicoloau, N.; Aarum, J.; Safari, H.; Muradrasoli, S.; Albert, J.; Högberg, B.; et al. Massive and rapid COVID-19 testing is feasible by extraction-free SARS-CoV-2 RT-qPCR 2020. medRxiv 2020. [Google Scholar]
- Brown, J.R.; Atkinson, L.; Shah, D.; Harris, K. Validation of an extraction-free RT-PCR protocol for detection of SARS-CoV2 RNA 2020. medRxiv 2020. [Google Scholar]
- Hasan, M.R.; Mirza, F.; Al-Hail, H.; Sundararaju, S.; Xaba, T.; Iqbal, M.; Alhussain, H.; Yassine, H.M.; Lopez, A.P.; Tang, P. Detection of SARS-CoV-2 RNA by direct RT-qPCR on nasopharyngeal specimens without extraction of viral RNA 2020. medRxiv 2020. [Google Scholar]
- Fomsgaard, A.S.; Rosenstierne, M.W. An alternative workflow for molecular detection of SARS-CoV-2 – escape from the NA extraction kit-shortage, Copenhagen, Denmark, March 2020. Eurosurveillance 2020, 25, 2000398. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Benchtop Thermocycler | Portable Thermocycler | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PCR | Mastermix Volume per Reaction (µL) | Mastermix Used | Template | Matrix | LoD (Cq Mean ± S.D) | PCR Efficiency (%) | R2 | LoD (Cq Mean ± S.D) | PCR Efficiency (%) | R2 |
| RT-qPCR | 20 | Invitrogen SuperScript™ III Platinum™ One-Step RT-qPCR Kit | ssRNA | Water | 120 (40.67 ± 0.29) | 84.30 | 0.9994 | 120 (38.70 ± 0.10) | 86.26 | 0.9985 |
| Sputum | No amplification | No amplification | ||||||||
| Nasal Exudate | No amplification | No amplification | ||||||||
| DIRECT-PCR | 20 | VitaNavi Direct One-Step S/P RT-qPCR TaqProbe Kit | ssRNA | Water | 120 (38.48 ± 0.57) | 84.94 | 0.9884 | 120 (36.99^) | 88.35 | 0.9729 |
| Sputum | 12 (38.79^) | 82.62 | 0.9944 | 12 (38.10^) | 88.64 | 0.9924 | ||||
| Nasal Exudate | 12 (38.72^) | 81.27 | 0.9986 | 1200 (36.47 ± 0.23) | 77.45 | 0.9976 | ||||
| Fast DIRECT-PCR | 10 | VitaNavi Direct One-Step S/P RT-qPCR TaqProbe Kit | ssRNA | Water | 600 (39.42^) | 81.72 | 0.9859 | 600 (36.63^) | 89.37 | 0.9639 |
| Sputum | 6 (39.28^) | 76.03 | 0.9824 | 600 (36.25 ± 0.46) | 85.52 | 0.9784 | ||||
| Nasal Exudate | 60 (39.34^) | 69.23 | 0.9865 | 60 (36.90^) | 81.08 | 0.9775 | ||||
| Plasmid | Water | 2 (39.75^) | 119.25 | 0.9896 | 20 (36.56 ± 0.26) | 113.17 | 0.9669 | |||
| Sputum | 2 (38.93^) | 101.91 | 0.9932 | 20 (35.40 ± 1.35) | 100.25 | 0.9756 | ||||
| Nasal Exudate | 20 (38.02 ± 0.55) | 96.72 | 0.9784 | 20 (36.66 ± 0.08) | 107.33 | 0.9931 | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wee, S.K.; Sivalingam, S.P.; Yap, E.P.H. Rapid Direct Nucleic Acid Amplification Test without RNA Extraction for SARS-CoV-2 Using a Portable PCR Thermocycler. Genes 2020, 11, 664. https://doi.org/10.3390/genes11060664
Wee SK, Sivalingam SP, Yap EPH. Rapid Direct Nucleic Acid Amplification Test without RNA Extraction for SARS-CoV-2 Using a Portable PCR Thermocycler. Genes. 2020; 11(6):664. https://doi.org/10.3390/genes11060664
Chicago/Turabian StyleWee, Soon Keong, Suppiah Paramalingam Sivalingam, and Eric Peng Huat Yap. 2020. "Rapid Direct Nucleic Acid Amplification Test without RNA Extraction for SARS-CoV-2 Using a Portable PCR Thermocycler" Genes 11, no. 6: 664. https://doi.org/10.3390/genes11060664
APA StyleWee, S. K., Sivalingam, S. P., & Yap, E. P. H. (2020). Rapid Direct Nucleic Acid Amplification Test without RNA Extraction for SARS-CoV-2 Using a Portable PCR Thermocycler. Genes, 11(6), 664. https://doi.org/10.3390/genes11060664