Comparative Pathway Integrator: A Framework of Meta-Analytic Integration of Multiple Transcriptomic Studies for Consensual and Differential Pathway Analysis

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Workflow of Comparative Pathway Integrator (CPI)
2.2. Meta-Analytic Pathway Analysis
2.3. Pathway Clustering for Reducing Redundancy and Enhancing Interpretation
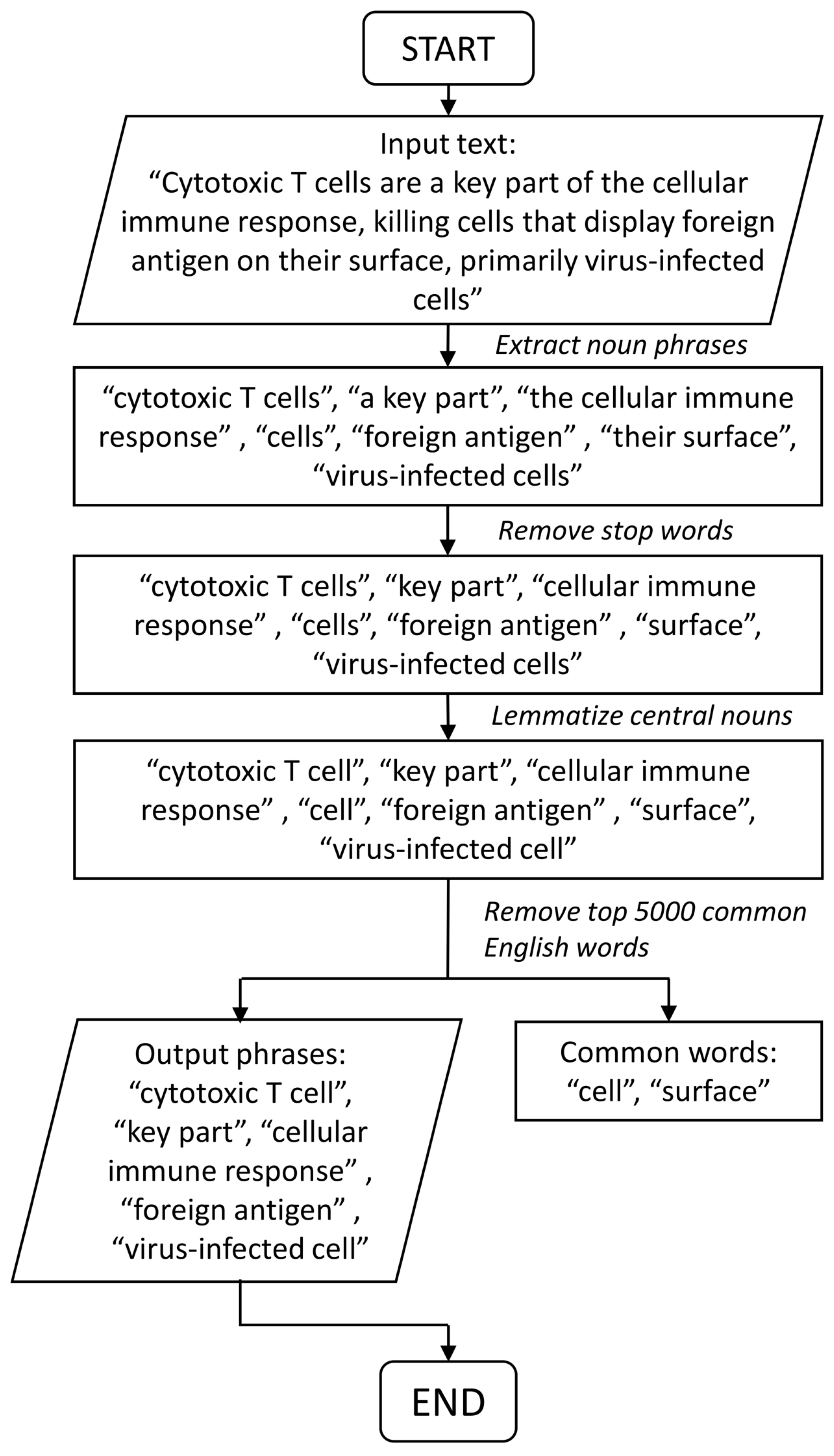
2.4. Text Mining for Automated Annotation and Knowledge Retrieval of Pathway Clusters
2.4.1. Motivation and Problem Setting
2.4.2. Pathway-Phrase Matrix
2.4.3. Test Statistics for Noun Phrase Enrichment Analysis
2.4.4. Permutation Test
2.4.5. Graphical and Spreadsheet Output
2.4.6. Datasets and Databases
3. Results
3.1. Application to Transcriptomic Data of Multiple Psychiatric Disorders
3.2. Justification to Penalize Pathway Description by Length in Text Mining
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R.; Jassal, B.; Jupe, S.; Korninger, F.; McKay, S.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2015, 44, D481–D487. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The molecular signatures database hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Rodchenkov, I.; Babur, O.; Luna, A.; Aksoy, B.A.; Wong, J.V.; Fong, D.; Franz, M.; Siper, M.C.; Cheung, M.; Wrana, M.; et al. Pathway Commons 2019 Update: Integration, analysis and exploration of pathway data. Nucleic Acids Res. 2020, 48, D489–D497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatri, P.; Sirota, M.; Butte, A.J. Ten years of pathway analysis: Current approaches and outstanding challenges. PLoS Comput. Biol. 2012, 8, e1002375. [Google Scholar] [CrossRef] [PubMed]
- Tseng, G.C.; Ghosh, D.; Feingold, E. Comprehensive literature review and statistical considerations for microarray meta-analysis. Nucleic Acids Res. 2012, 40, 3785–3799. [Google Scholar] [CrossRef] [Green Version]
- Shen, K.; Tseng, G.C. Meta-analysis for pathway enrichment analysis when combining multiple genomic studies. Bioinformatics 2010, 26, 1316–1323. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Tagett, R.; Donato, M.; Mitrea, C.; Draghici, S. A novel bi-level meta-analysis approach: Applied to biological pathway analysis. Bioinformatics 2016, 32, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Mitrea, C.; Tagett, R.; Draghici, S. DANUBE: Data-driven meta-ANalysis using UnBiased empirical distributions—Applied to biological pathway analysis. Proc. IEEE 2016, 105, 496–515. [Google Scholar] [CrossRef]
- Plaisier, S.B.; Taschereau, R.; Wong, J.A.; Graeber, T.G. Rank–rank hypergeometric overlap: Identification of statistically significant overlap between gene-expression signatures. Nucleic Acids Res. 2010, 38, e169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahill, K.M.; Huo, Z.; Tseng, G.C.; Logan, R.W.; Seney, M.L. Improved identification of concordant and discordant gene expression signatures using an updated rank-rank hypergeometric overlap approach. Sci. Rep. 2018, 8, 9588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tseng, G.C. An adaptively weighted statistic for detecting differential gene expression when combining multiple transcriptomic studies. Ann. Appl. Stat. 2011, 5, 994–1019. [Google Scholar] [CrossRef]
- Tseng, G.C.; Wong, W.H. Tight clustering: A resampling-based approach for identifying stable and tight patterns in data. Biometrics 2005, 61, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, T.; Huo, Z.; Kuo, A.; Zhu, L.; Fang, Z.; Zeng, X.; Lin, C.W.; Liu, S.; Wang, L.; Liu, P.; et al. MetaOmics: Analysis pipeline and browser-based software suite for transcriptomic meta-analysis. Bioinformatics 2019, 35, 1597–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarca, A.L.; Bhatti, G.; Romero, R. A comparison of gene set analysis methods in terms of sensitivity, prioritization and specificity. PLoS ONE 2013, 8, e79217. [Google Scholar] [CrossRef]
- Bayerlová, M.; Jung, K.; Kramer, F.; Klemm, F.; Bleckmann, A.; Beißbarth, T. Comparative study on gene set and pathway topology-based enrichment methods. BMC Bioinform. 2015, 16, 334. [Google Scholar] [CrossRef] [Green Version]
- Huo, Z.; Tang, S.; Park, Y.; Tseng, G. p-value evaluation, variability index and biomarker categorization for adaptively weighted Fisher’s meta-analysis method in omics applications. Bioinformatics 2019, 36, 524–532. [Google Scholar] [CrossRef] [Green Version]
- Viera, A.J.; Garrett, J.M. Understanding interobserver agreement: The kappa statistic. Fam. Med. 2005, 37, 360–363. [Google Scholar]
- Monti, S.; Tamayo, P.; Mesirov, J.; Golub, T. Consensus clustering: A resampling-based method for class discovery and visualization of gene expression microarray data. Mach. Learn. 2003, 52, 91–118. [Google Scholar] [CrossRef]
- Maitra, R.; Ramler, I.P. Clustering in the Presence of Scatter. Biometrics 2009, 65, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, G.C. Penalized and weighted K-means for clustering with scattered objects and prior information in high-throughput biological data. Bioinformatics 2007, 23, 2247–2255. [Google Scholar] [CrossRef]
- Rousseeuw, P.J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Benoit, K.; Matsuo, A.; Benoit, M.K. R Package: ‘spacyr’. 2018. Available online: https://cran.r-project.org/web/packages/spacyr/spacyr.pdf (accessed on 25 February 2020).
- Honnibal, M.; Montani, I. spaCy 2: Natural language understanding with Bloom embeddings, convolutional neural networks and incremental parsing. To Appear 2017, 7. [Google Scholar]
- Feinerer, I. Introduction to the tm Package Text Mining in R. 2013. Available online: https://cran.r-project.org/web/packages/tm/tm.pdf (accessed on 25 February 2020).
- Rinker, T. R Package: ‘textstem’. 2018. Available online: https://cran.r-project.org/web/packages/textstem/textstem.pdf (accessed on 25 February 2020).
- Word Frequency Data. Top 5000 common English Words. 2017. Available online: http://www.wordfrequency.info (accessed on 25 February 2020).
- Feinerer, I.; Hornik, K.; Wallace, M.; Hornik, M.K. Package ‘wordnet’. 2017. Available online: https://cran.r-project.org/web/packages/wordnet/wordnet.pdf (accessed on 25 February 2020).
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Arion, D.; Huo, Z.; Enwright, J.F.; Corradi, J.P.; Tseng, G.; Lewis, D.A. Transcriptome alterations in prefrontal pyramidal cells distinguish schizophrenia from bipolar and major depressive disorders. Biol. Psychiatry 2017, 82, 594–600. [Google Scholar] [CrossRef]
- Bousman, C.A.; Chana, G.; Glatt, S.J.; Chandler, S.D.; Lucero, G.R.; Tatro, E.; May, T.; Lohr, J.B.; Kremen, W.S.; Tsuang, M.T.; et al. Preliminary evidence of ubiquitin proteasome system dysregulation in schizophrenia and bipolar disorder: Convergent pathway analysis findings from two independent samples. Am. J. Med. Genet. Part Neuropsychiatr. Genet. 2010, 153, 494–502. [Google Scholar] [CrossRef] [Green Version]
- Arion, D.; Corradi, J.P.; Tang, S.; Datta, D.; Boothe, F.; He, A.; Cacace, A.M.; Zaczek, R.; Albright, C.F.; Tseng, G.; et al. Distinctive transcriptome alterations of prefrontal pyramidal neurons in schizophrenia and schizoaffective disorder. Mol. Psychiatry 2015, 20, 1397–1405. [Google Scholar] [CrossRef]
- Sheng, Z.H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [Green Version]
- Darby, M.; Yolken, R.H.; Sabunciyan, S. Consistently altered expression of gene sets in postmortem brains of individuals with major psychiatric disorders. Transl. Psychiatry 2016, 6, e890. [Google Scholar] [CrossRef] [PubMed]
- Gandal, M.J.; Haney, J.R.; Parikshak, N.N.; Leppa, V.; Ramaswami, G.; Hartl, C.; Schork, A.J.; Appadurai, V.; Buil, A.; Werge, T.M.; et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 2018, 359, 693–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanz, T.A.; Reinhart, V.; Sheehan, M.J.; Rizzo, S.J.S.; Bove, S.E.; James, L.C.; Volfson, D.; Lewis, D.A.; Kleiman, R.J. Postmortem transcriptional profiling reveals widespread increase in inflammation in schizophrenia: A comparison of prefrontal cortex, striatum, and hippocampus among matched tetrads of controls with subjects diagnosed with schizophrenia, bipolar or major depressive disorder. Transl. Psychiatry 2019, 9, 1–13. [Google Scholar]
- Ramaker, R.C.; Bowling, K.M.; Lasseigne, B.N.; Hagenauer, M.H.; Hardigan, A.A.; Davis, N.S.; Gertz, J.; Cartagena, P.M.; Walsh, D.M.; Vawter, M.P.; et al. Post-mortem molecular profiling of three psychiatric disorders. Genome Med. 2017, 9, 72. [Google Scholar] [CrossRef] [Green Version]
- McGrath, L.M.; Cornelis, M.C.; Lee, P.H.; Robinson, E.B.; Duncan, L.E.; Barnett, J.H.; Huang, J.; Gerber, G.; Sklar, P.; Sullivan, P.; et al. Genetic predictors of risk and resilience in psychiatric disorders: A cross-disorder genome-wide association study of functional impairment in major depressive disorder, bipolar disorder, and schizophrenia. Am. J. Med Genet. Part Neuropsychiatr. Genet. 2013, 162, 779–788. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
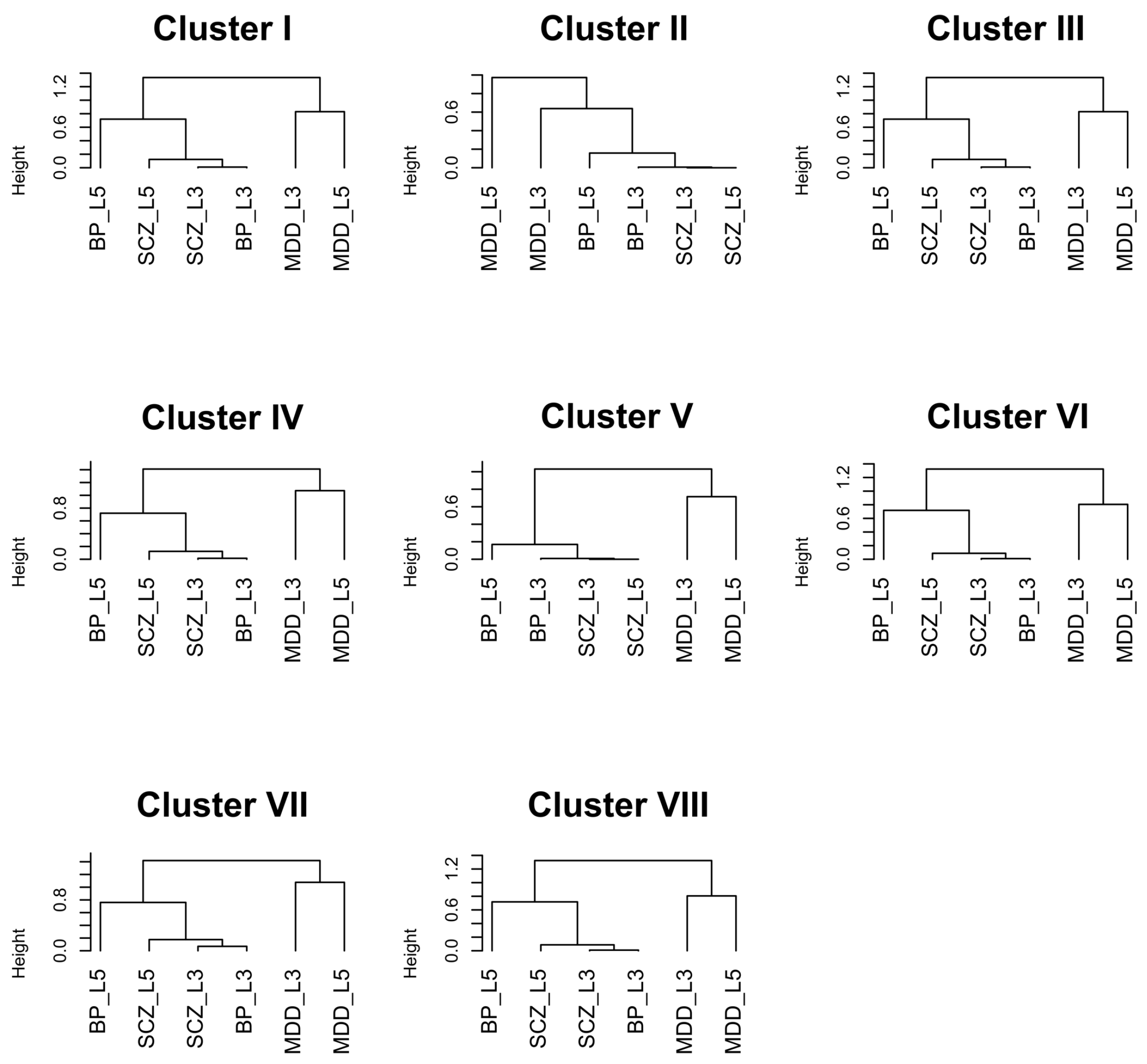
| Cluster | Keywords |
|---|---|
| I | insulin, NGF, focal adhesion, BDNF, neurotrophins, Trk tyrosine kinase receptor, insulin receptor substrate, insulin receptor tyrosine kinase, Ras MAPK pathway, FAK |
| II | neuron |
| III | transcription, nucleoplasm, chromosome, nuclear content, nucleolus, RNA |
| IV | metabolism, mRNA, ribosome, replication, chemical reaction, cRNA, vRNA, viral protein, NUMB, nucleus |
| V | cell death, apoptotic process, activation, endogenous cellular process, programmed cell death, apoptosis |
| VI | mitochondrion, organelle, mitochondrial envelope, organelle envelope, lipid bilayer, inner elumen facing lipid bilayer, semiautonomous self replicating organelle, tissue respiration, virtually eukaryotic cell, cytoplasm |
| VII | degradation, APC/C, apoptosis, CDC20, CDH1, mitotic protein, MHC, multiubiquitination, ubiquitin 26s proteasome system, exogenous antigen |
| VIII | respiratory electron transport, ATP synthesis, inner mitochondrial membrane, chemiosmotic gradient, brown fat, rotenone, FAD, mitochondrial matrix, body temperature, NAD |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, X.; Zong, W.; Lin, C.-W.; Fang, Z.; Ma, T.; Lewis, D.A.; Enwright, J.F.; Tseng, G.C. Comparative Pathway Integrator: A Framework of Meta-Analytic Integration of Multiple Transcriptomic Studies for Consensual and Differential Pathway Analysis. Genes 2020, 11, 696. https://doi.org/10.3390/genes11060696
Zeng X, Zong W, Lin C-W, Fang Z, Ma T, Lewis DA, Enwright JF, Tseng GC. Comparative Pathway Integrator: A Framework of Meta-Analytic Integration of Multiple Transcriptomic Studies for Consensual and Differential Pathway Analysis. Genes. 2020; 11(6):696. https://doi.org/10.3390/genes11060696
Chicago/Turabian StyleZeng, Xiangrui, Wei Zong, Chien-Wei Lin, Zhou Fang, Tianzhou Ma, David A. Lewis, John F. Enwright, and George C. Tseng. 2020. "Comparative Pathway Integrator: A Framework of Meta-Analytic Integration of Multiple Transcriptomic Studies for Consensual and Differential Pathway Analysis" Genes 11, no. 6: 696. https://doi.org/10.3390/genes11060696
APA StyleZeng, X., Zong, W., Lin, C. -W., Fang, Z., Ma, T., Lewis, D. A., Enwright, J. F., & Tseng, G. C. (2020). Comparative Pathway Integrator: A Framework of Meta-Analytic Integration of Multiple Transcriptomic Studies for Consensual and Differential Pathway Analysis. Genes, 11(6), 696. https://doi.org/10.3390/genes11060696