Classification of Microarray Gene Expression Data Using an Infiltration Tactics Optimization (ITO) Algorithm


Abstract
:1. Introduction
- Find: In this stage, the LIG members analyze the field position to make a strategy and find the most appropriate target to attack.
- Fix: In this stage, the LIG members use different light-weight weapons to infiltrate into enemy areas.
- Flank/Fight: In this stage, LIG members keep the enemy pinned down so they could not reorganize their forces while the FT performs a detailed offensive in the area independently.
- Finish: In this stage, the FT members apply heavier weapons to cleanup the area and gain full control over the enemy.
2. Background and Literature Review
2.1. Phases of Microarray Gene Expression Data
2.1.1. Phase 1: Pre-Processing
2.1.2. Phase 2: Feature Subset Selection
2.1.3. Phase 3: Learning and Classification
2.2. Literature Review
2.2.1. Microarray Quality Control (MAQC)
2.2.2. Feature Selection Algorithms
2.2.3. Ensemble Based Approaches
2.2.4. Heterogeneous Ensemble Classifiers
2.2.5. Bio-Inspired Algorithms
2.2.6. Deep Learning Based Approaches
2.2.7. Image Based Cancer Classification
2.2.8. Cancer Detection Using Transfer Learning
2.2.9. Summary of Literature Review
3. Proposed Algorithm
- Find: In this stage, a random grid search is applied on the 4-dimensional search space comprising of pre-processing methods, FSS methods, Subset sizes, and Validation methods to generate “attack vectors” (tuples of length 4 each from the search space with different combinations) for LIG and FT members e.g., (Quantile method, mRMR, 50 features, 10 Fold CV) is one such tuple. Details of the options used for each of these dimensions are given below.
- Fix: In this stage, each of the LIG members use one of the attack vectors to construct individual models. An efficiency index is calculated using Matthews Correlation Coefficient (MCC) and average classification accuracy (score) as:where LIG(i) is the i-th member of LIG. Similar to MAQC-II benchmarks, MCC and accuracy are used to compute . In addition, our analysis from earlier experimentation showed that, in the case of overfitting, though the average accuracy/score of the model seemingly improves but simultaneously the MCC of the model decreases. Hence, these two measures were used to decide the trade-off between accuracy and MCC at the time of base classifier selection. The value of MCC ranges between −1 and +1, but, for our experimentation, we used only (0, 1] or MCC > 0 i.e., anything better than random guess. The accuracy ranges between [0, 1] range. Both measures are equally important, thus we use product as a statistical conjunction function. It helps balance the trade-offs between MCC and Accuracy. In our experiments, we observed that helped in improving both the MCC and accuracy in some cases and helped achieve a good trade-off between MCC and accuracy in other cases. A fitness threshold is used to filter in “successful” members from the whole LIG i.e.,The value of is chosen such that it filters at least the top 33% of the LIG members for . Once the ensemble is formed, the value of can be adjusted to tune the ensemble for maximum yield as explained below.
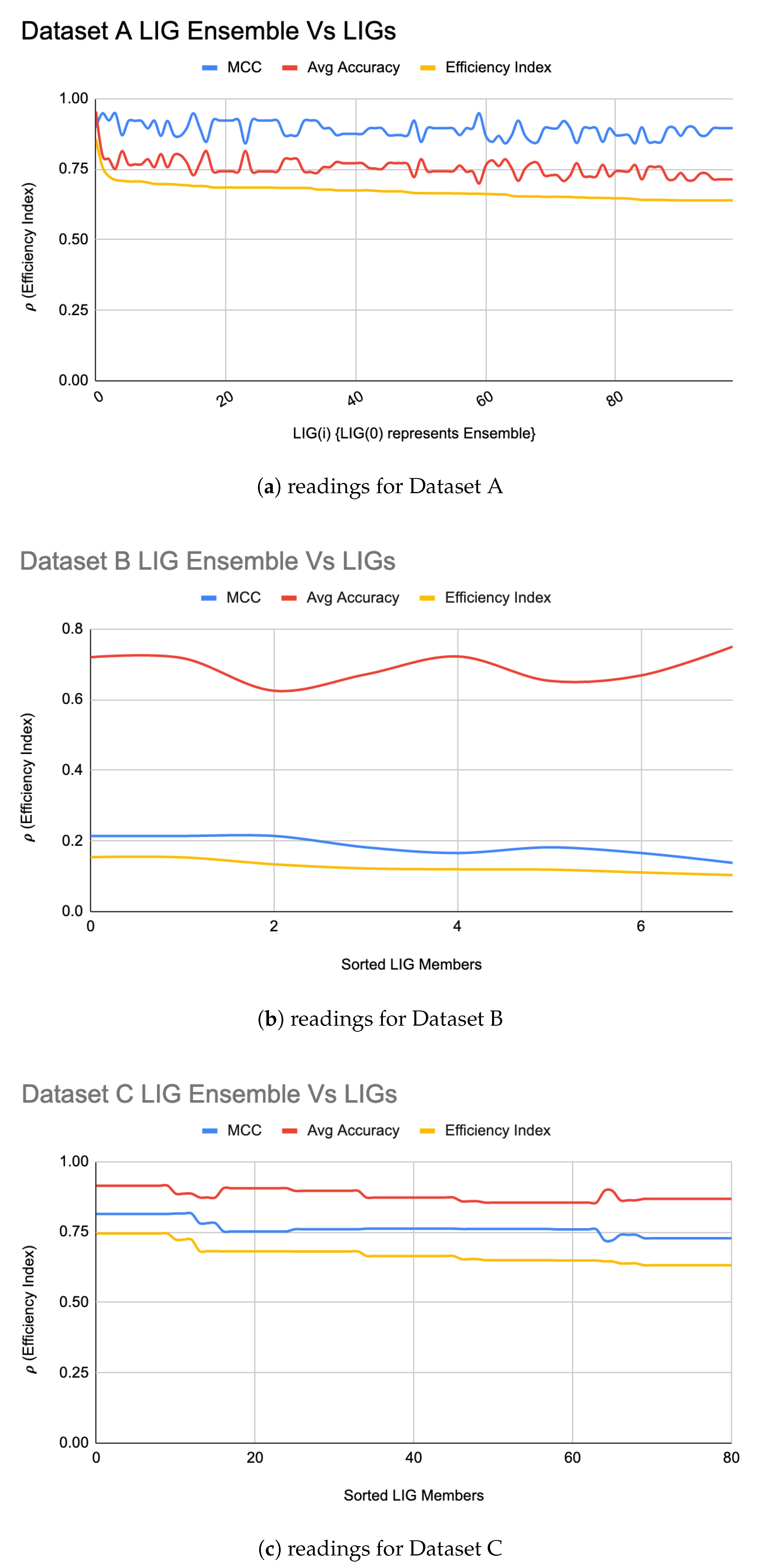
- Flank/Fight: In this stage, a heterogeneous ensemble of a subset of “successful” LIG members (MCC > 0) is formed such that:The ensemble is formed iteratively using a majority-vote method. In each iteration, the top LIG(i) is added to the and is computed to ensure that the newly added LIG(i) did not deteriorate the ensemble performance. If an LIG(i) causes decline in the , it is discarded.The takes relatively very short time to build while each FT(i) may take several hours to days to train (depending upon the parameter-space), thus, for the time-sensitive cases e.g., in the domain of pandemic diseases where an early prediction may be required, can be used until FT(i) are being trained. When the FT(i) are trained and the ensemble updated for improved performance, a follow-up prediction could be done which will either strengthen the confidence in prediction if both and agree on the prediction Or could be used to over-ride the earlier prediction.
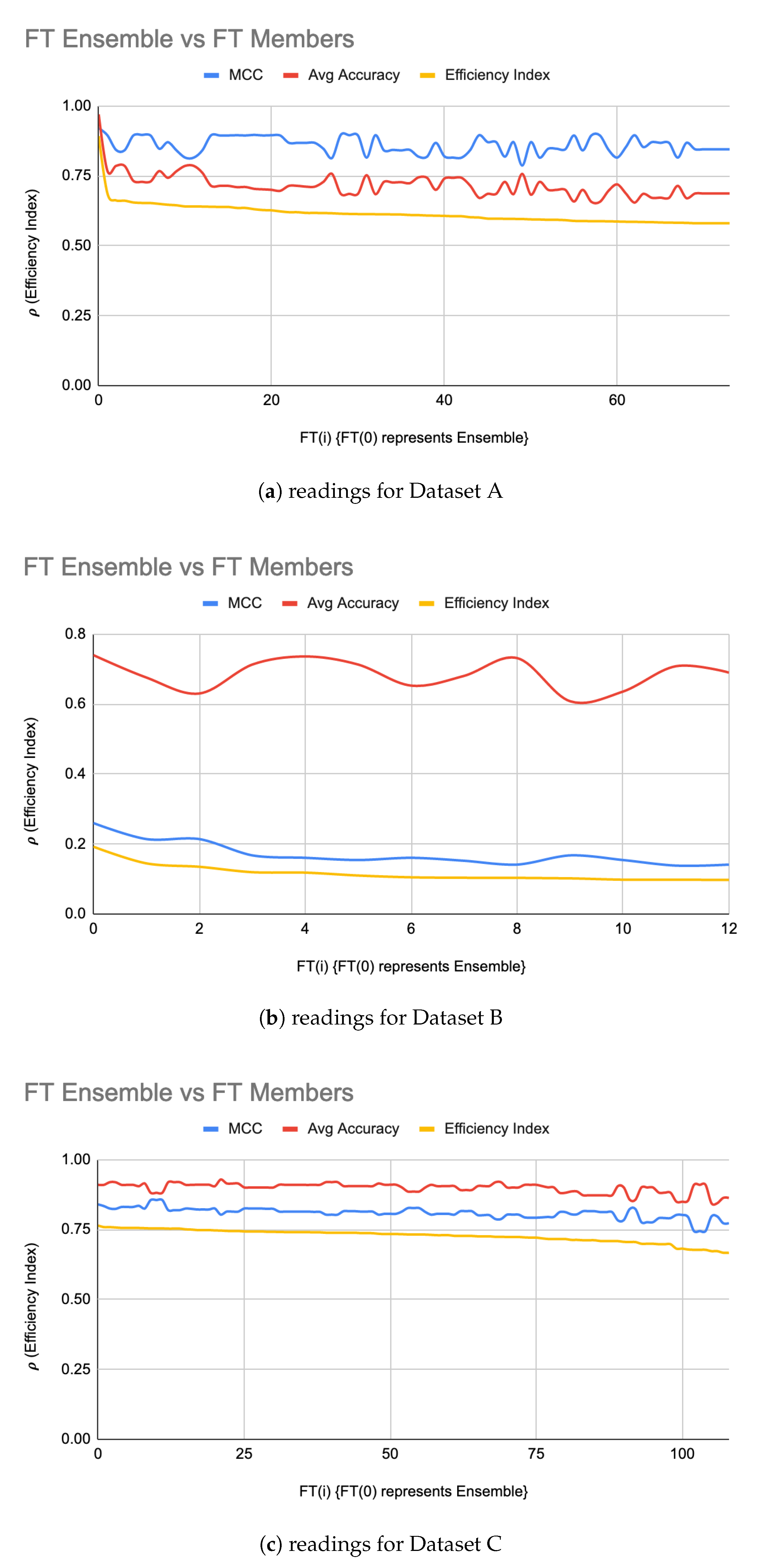
- Finish: In this stage, the FT members apply advanced classifiers such as Deep Neural Networks, SVM, etc. to build fine-tuned models. The “successful” FT members are filtered in using:where FT(i) is the ith member of FT. A fitness threshold is used to filter in “successful” FT members i.e.,Then, is computed from FT subsets such that:Finally, a is formed using filtered-in LIG(i) and filtered-in FT(i). The following different approaches can be used to build the :
- (a)
- simply combine all the LIG and FT members from & , respectively. However, through empirical analysis, it was found that this approach actually causes a decline in MCC and/or average accuracy of the model.
- (b)
- start with one of or and call it . Choose base classifiers from the other ensemble with and add to . However, starting with an ensemble with higher would cause all of them to fail on , thus resulting in no further improvement. In addition, our experiments showed that, starting with an ensemble with lower , the optimization gain was not as good as the next approach because the condition filtered out many classifiers which still could help with reducing misclassifications of ensembles hence improve both the accuracy and MCC.
- (c)
- rebuild the from scratch using LIG(i) ∪ FT(i) ordered by . This approach was found effective to further enhance the performance.
Pseudo Code
| Algorithm 1: ITO Algorithm |
| input: |
| T: t × f matrix - training dataset with t samples and f features; |
| V: v × f matrix - validation dataset - with v samples and f features; |
| preps={Imputer, Robust, Quantile, Standard, …} - set of preprocessing methods; |
| searchRadius={10, 50, 100, 150, 200, 250, …} - set of FSS sizes; |
| searchStrategy={JMI, JMIM, mRMR …} - set of FSS methods; |
| successEvaluation={10 Fold CV, LOOCV, …} - set of validation methods; |
| ={DT, AdaBoost, Extra Tree, …} - set of parameter-free classifiers; |
| ={DNN, SVM, Random Forest, …} - set of parameterized classifiers |
| output: |
| BEGIN |
| G ← GenerateOptionsGrid(searchRadius, searchStrategy, successEvaluation, preps); |
| Choose ⊂ G using Randomized Grid Search; |
| ← (T, V, , ) //Algorithm 2; |
| Choose ⊂ G using Randomized Grid Search; |
| ← (T, V, , ) //Algorithm 3; |
| return ; |
| END |
| Algorithm 2: Compute |
 |
| Algorithm 3: Compute |
 |
4. Experimental Setup
4.1. Benchmark Datasets
4.2. Description of Datasets Used
4.3. FT Parameter Setting
- learning_rate: [“constant”, “invscaling”, “adaptive”]
- alpha: [1, 0.1, 0.001, 0.0001, 0.00001, 0.000001, 0.0000001, 0.00000001]
- activation: [“logistic”, “relu”, “tanh”]
- hidden_layer_sizes: [(100,1), (100,2), (100,3)]
- C = [0.001, 0.01, 0.1, 1, 10]
- gamma = [0.001, 0.01, 0.1, 1]
- kernels = [’rbf’,’linear’]
- estimators: [100, 300, 500, 800, 1000]
- criterion: [‘gini’, ‘entropy’]
- bootstrap: [True, False]
4.4. ITO Parameter Setting
5. Results and Analysis
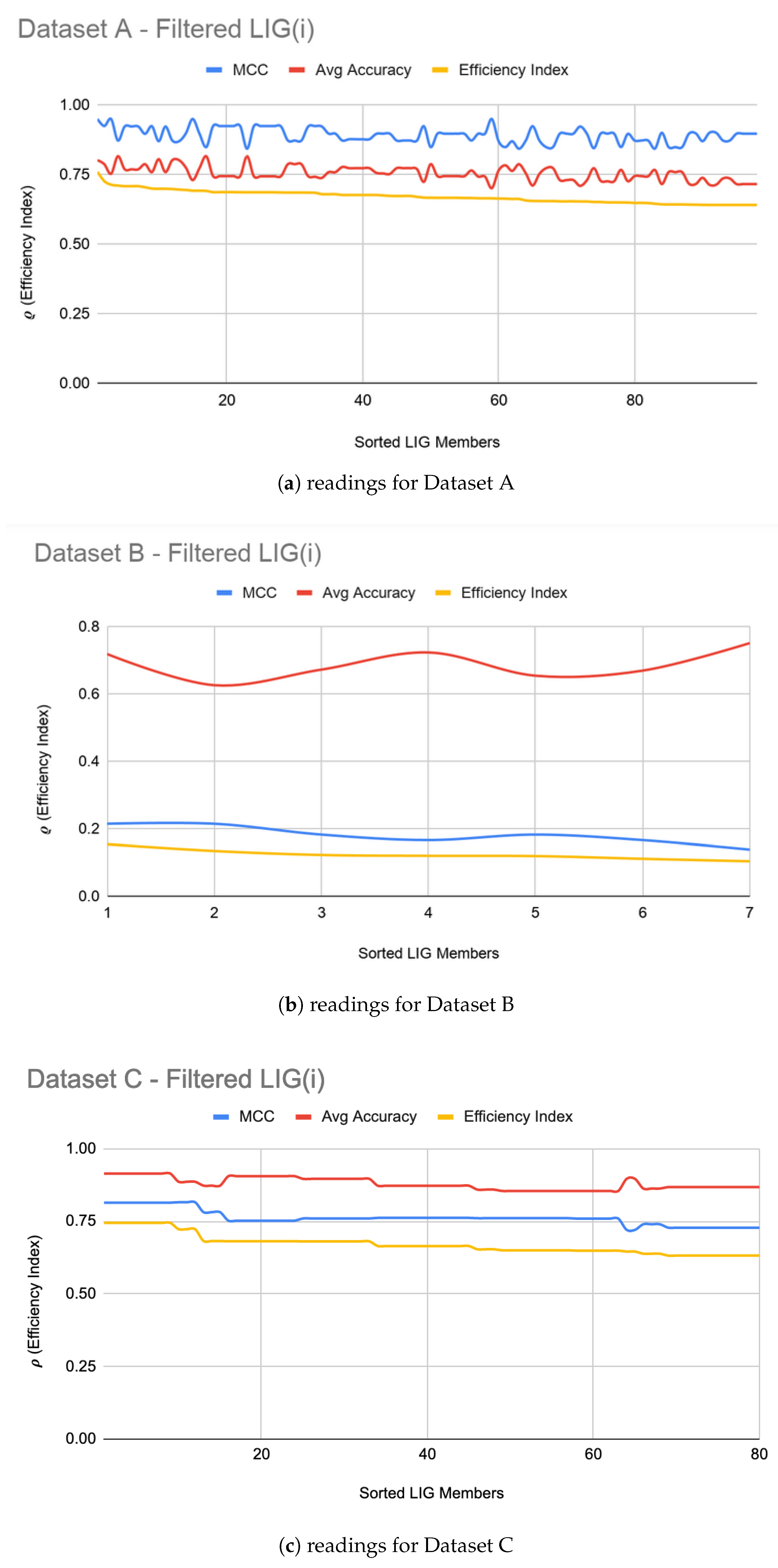
5.1. LIG Optimizations
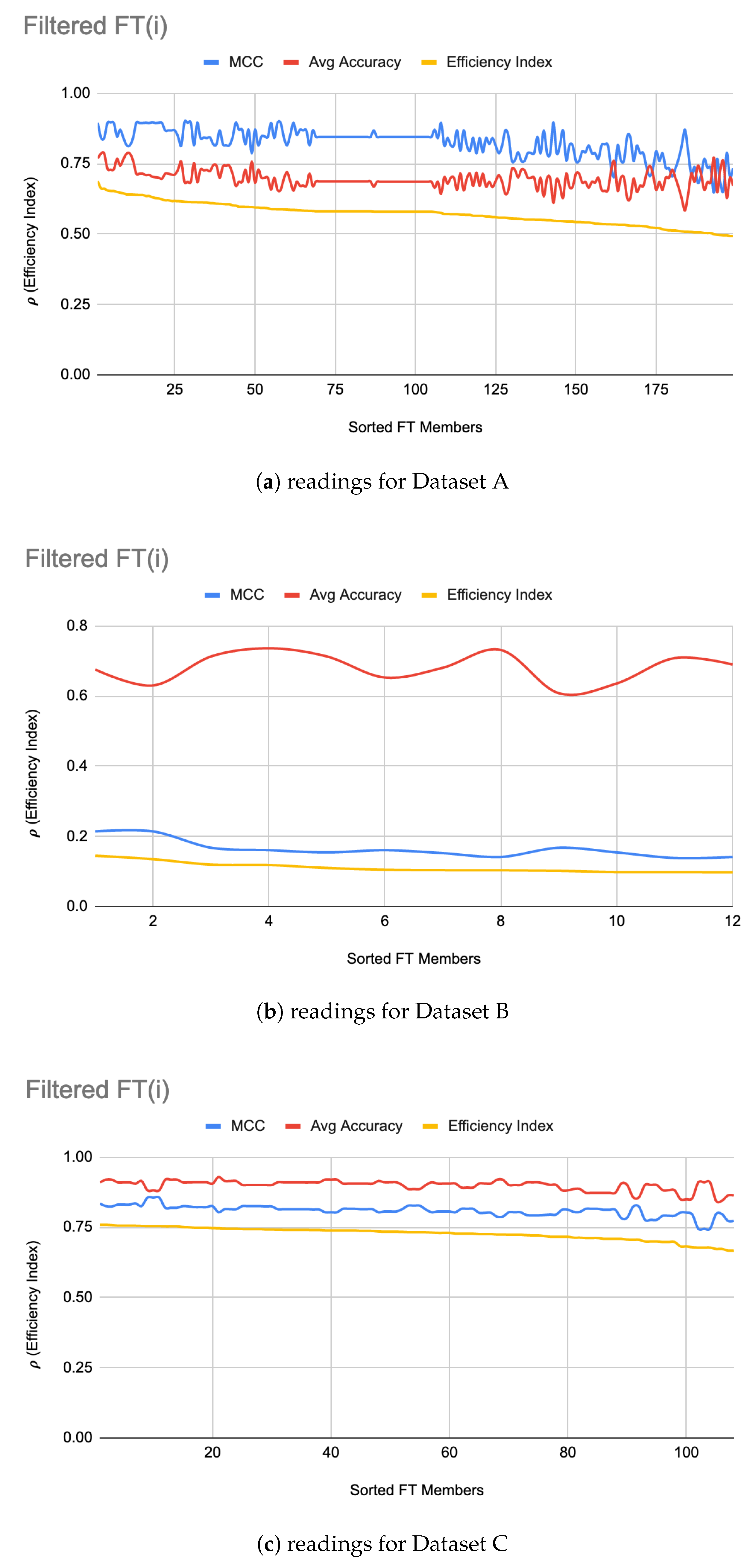
5.2. FT Optimizations
6. Machine Specifications
7. Time Complexity
- number of samples (t)
- number of features (f)
- number of different preprocessing methods (P) and maximum execution time for preprocessing () of dataset of size txf
- number of FSS methods (FM) and maximum execution time for feature selection () from f features. This is one of the most time-consuming steps of the algorithm because the underlying methods need to calculate pair-wise mutual information for feature ranking, which is eventually used to pick the top features.
- Subset sizes (S)
- Validation methods (V)
- number of parameterized classifiers () and maximum time to train a parameterized classifier () This is the second most time-consuming step of the algorithm. Parameter tuning for the classifiers requires trying different combinations of parameter values and find the most effective one.
- number of non-parameterized (parameter-free) classifiers () and maximum time to train a parameter-free classifier ().
- Ensemble construction time (=FT Ensemble, =LIG Ensemble, =Overall Ensemble).
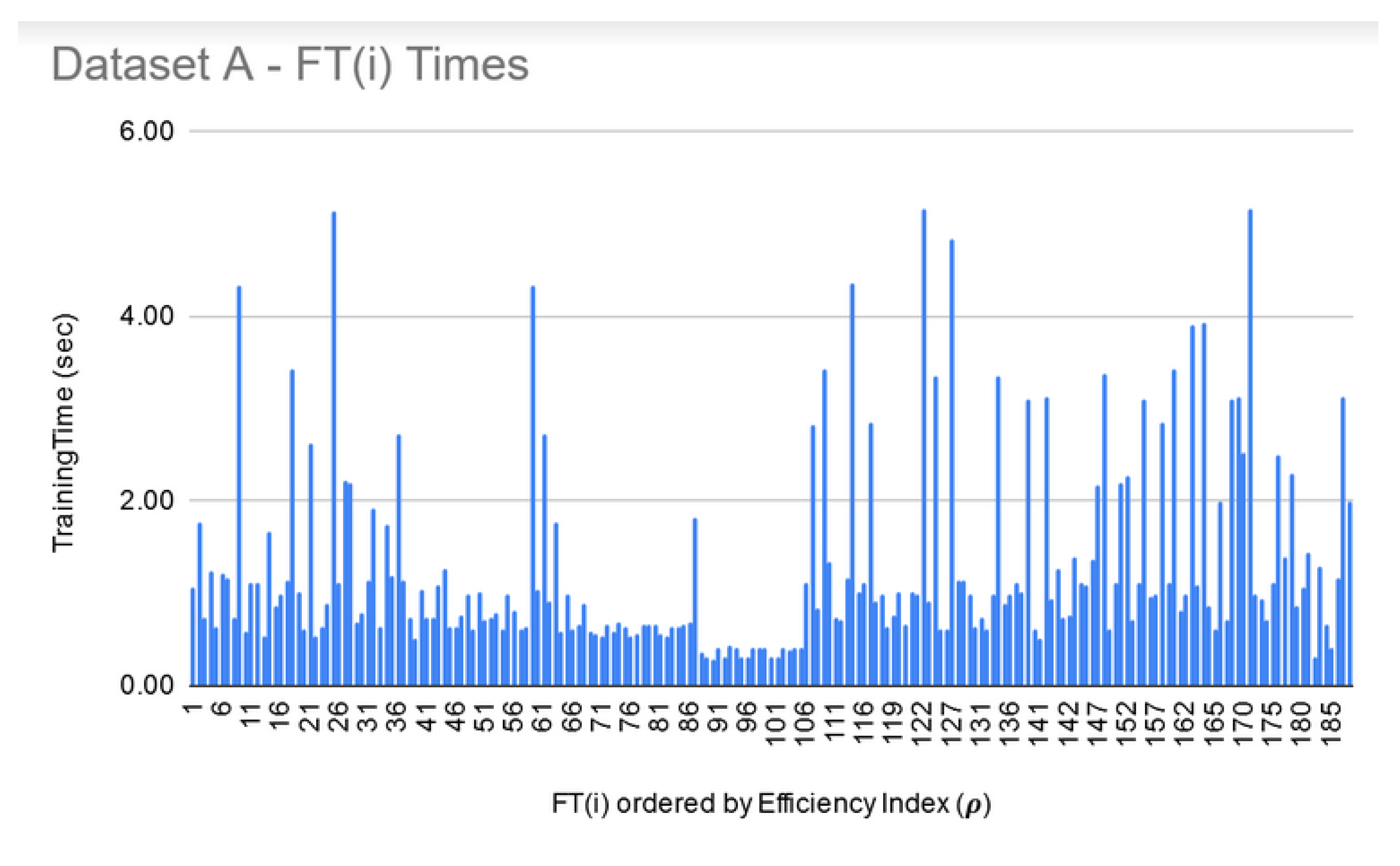
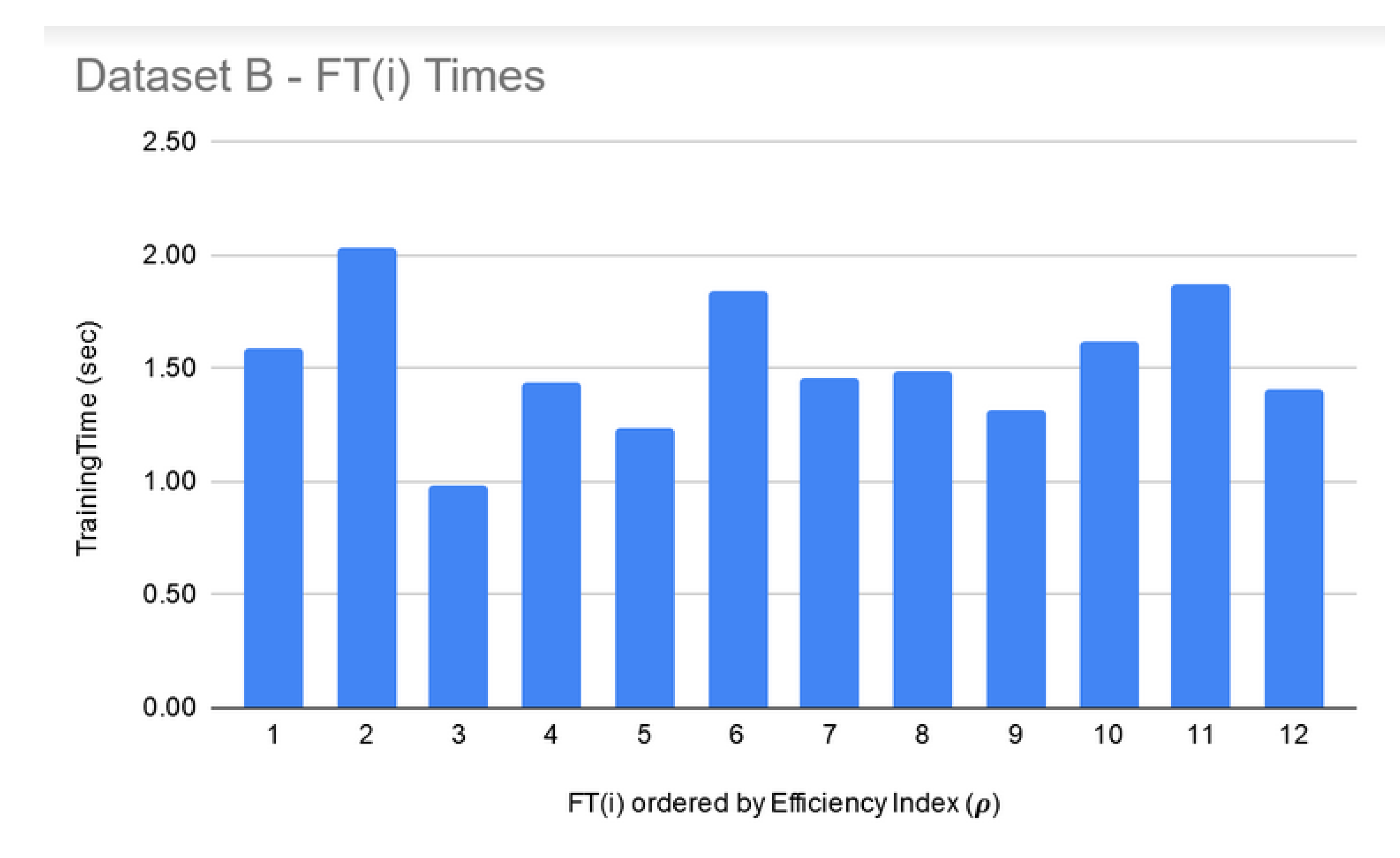
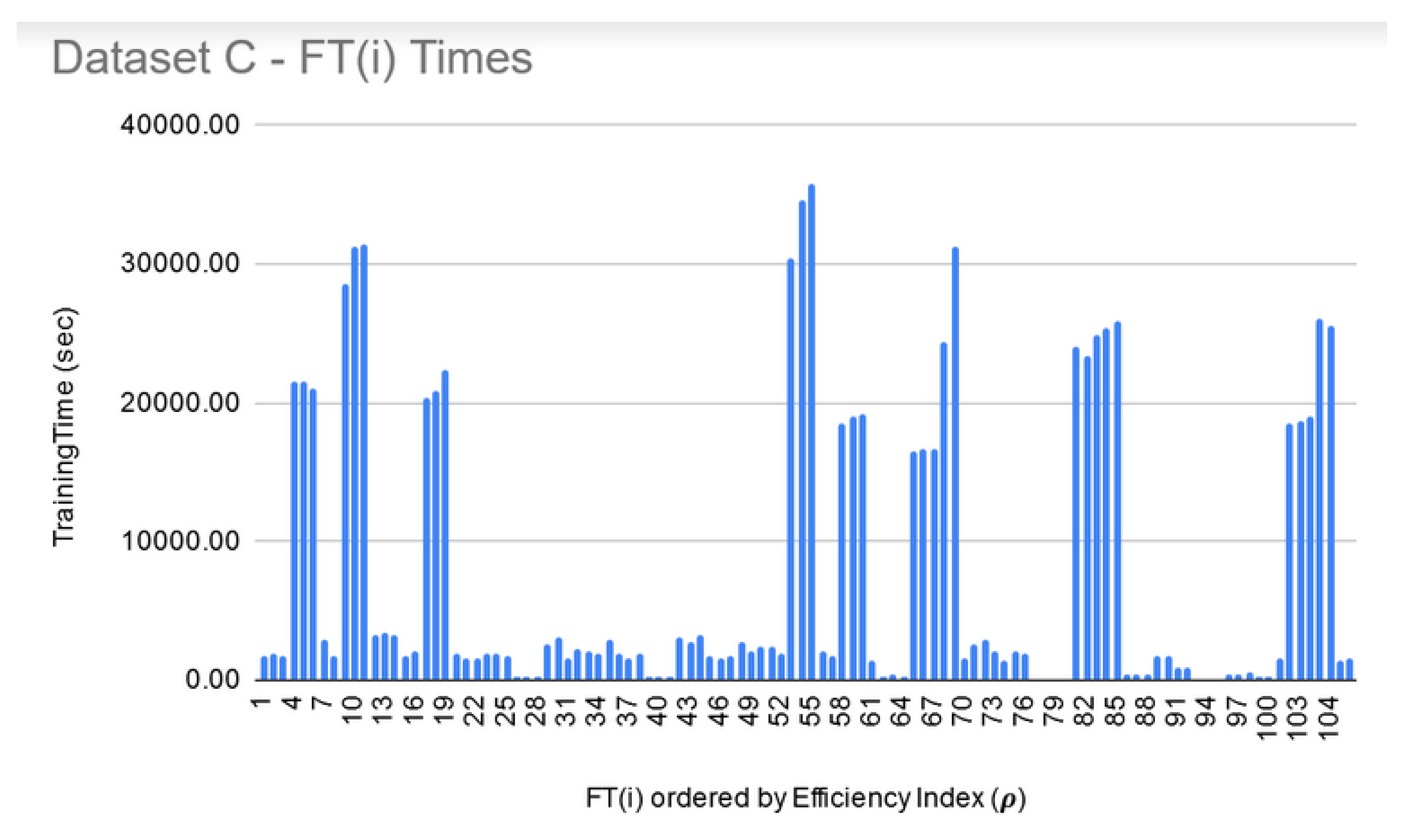
8. Execution Times
9. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ITO | Infiltration Tactics Optimization algorithm. |
| MAQC-II | Microarray Quality Control study Phase II |
| mRMR | Minimum Redundancy Maximum Relevance. |
| MCC | Matthews Correlation Coefficient |
| HAHR | High Accuracy, High Reliability |
References
- Alanni, R.; Hou, J.; Azzawi, H.; Xiang, Y. Deep gene selection method to select genes from microarray datasets for cancer classification. BMC Bioinform. 2019, 20, 608. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Morstatter, F.; Sharma, S.; Alelyani, S.; Anand, A.; Liu, H. Advancing feature selection research. ASU Feature Sel. Repos. 2010, 1–28. [Google Scholar]
- Elloumi, M.; Zomaya, A.Y. Algorithms in Computational Molecular Biology: Techniques, Approaches and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2011; Volume 21. [Google Scholar]
- Bolón-Canedo, V.; Sánchez-Marono, N.; Alonso-Betanzos, A.; Benítez, J.M.; Herrera, F. A review of microarray datasets and applied feature selection methods. Inf. Sci. 2014, 282, 111–135. [Google Scholar] [CrossRef]
- Almugren, N.; Alshamlan, H. A survey on hybrid feature selection methods in microarray gene expression data for cancer classification. IEEE Access 2019, 7, 78533–78548. [Google Scholar] [CrossRef]
- Ding, C.; Peng, H. Minimum redundancy feature selection from microarray gene expression data. J. Bioinform. Comput. Biol. 2005, 3, 185–205. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cheng, K.; Wang, S.; Morstatter, F.; Trevino, R.P.; Tang, J.; Liu, H. Feature selection: A data perspective. ACM Comput. Surv. (CSUR) 2017, 50, 94. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Long, F.; Ding, C. Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Fakoor, R.; Ladhak, F.; Nazi, A.; Huber, M. Using deep learning to enhance cancer diagnosis and classification. In Proceedings of the International Conference on Machine Learning, Atlanta, GA, USA, 16–21 June 2013; ACM: New York, NY, USA, 2013; Volume 28. [Google Scholar]
- Chen, Y.; Li, Y.; Narayan, R.; Subramanian, A.; Xie, X. Gene expression inference with deep learning. Bioinformatics 2016, 32, 1832–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevakula, R.K.; Singh, V.; Verma, N.K.; Kumar, C.; Cui, Y. Transfer learning for molecular cancer classification using deep neural networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 16, 2089–2100. [Google Scholar] [CrossRef]
- Shi, L.; Campbell, G.; Jones, W.D.; Campagne, F.; Wen, Z.; Walker, S.J.; Su, Z.; Chu, T.M.; Goodsaid, F.M.; Pusztai, L.; et al. The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nat. Biotechnol. 2010, 28, 827. [Google Scholar]
- Djebbari, A.; Culhane, A.C.; Armstrong, A.J.; Quackenbush, J. AI Methods for Analyzing Microarray Data; Dana-Farber Cancer Institute: Boston, MA, USA, 2007. [Google Scholar]
- Selvaraj, C.; Kumar, R.S.; Karnan, M. A survey on application of bio-inspired algorithms. Int. J. Comput. Sci. Inf. Technol. 2014, 5, 366–370. [Google Scholar]
- Duncan, J.; Insana, M.; Ayache, N. Biomedical Imaging and Analysis In the Age of Sparsity, Big Data, and Deep Learning. Proc. IEEE 2020, 108. [Google Scholar] [CrossRef]
- Bojarski, M.; Del Testa, D.; Dworakowski, D.; Firner, B.; Flepp, B.; Goyal, P.; Jackel, L.D.; Monfort, M.; Muller, U.; Zhang, J.; et al. End to end learning for self-driving cars. arXiv 2016, arXiv:1604.07316. [Google Scholar]
- Huynh, B.Q.; Li, H.; Giger, M.L. Digital mammographic tumor classification using transfer learning from deep convolutional neural networks. J. Med. Imaging 2016, 3, 034501. [Google Scholar] [CrossRef] [PubMed]
- Spanhol, F.A.; Oliveira, L.S.; Petitjean, C.; Heutte, L. Breast cancer histopathological image classification using Convolutional Neural Networks. In Proceedings of the 2016 International Joint Conference on Neural Networks (IJCNN), Vancouver, BC, Canada, 24–29 July 2016; pp. 2560–2567. [Google Scholar] [CrossRef]
- Han, Z.; Wei, B.; Zheng, Y.; Yin, Y.; Li, K.; Li, S. Breast cancer multi-classification from histopathological images with structured deep learning model. Sci. Rep. 2017, 7, 4172. [Google Scholar] [CrossRef] [PubMed]
- Lévy, D.; Jain, A. Breast mass classification from mammograms using deep convolutional neural networks. arXiv 2016, arXiv:1612.00542. [Google Scholar]
- Liao, Q.; Ding, Y.; Jiang, Z.L.; Wang, X.; Zhang, C.; Zhang, Q. Multi-task deep convolutional neural network for cancer diagnosis. Neurocomputing 2019, 348, 66–73. [Google Scholar] [CrossRef]
- Chapman, A. Digital Games as History: How Videogames Represent the Past and Offer Access to Historical Practice; Routledge Advances in Game Studies; Taylor & Francis: Abingdon, UK, 2016; p. 185. [Google Scholar]
- Ikeda, N.; Watanabe, S.; Fukushima, M.; Kunita, H. Itô’s Stochastic Calculus and Probability Theory; Springer: Tokyo, Japan, 2012. [Google Scholar]
- Sato, I.; Nakagawa, H. Approximation analysis of stochastic gradient Langevin dynamics by using Fokker–Planck equation and Ito process. In International Conference on Machine Learning; PMLR: Bejing, China, 2014; pp. 982–990. [Google Scholar]
- Polley, E.C.; Van Der Laan, M.J. Super Learner in Prediction. U.C. Berkeley Division of Biostatistics Working Paper Series. Working Paper 266. May 2010. Available online: https://biostats.bepress.com/ucbbiostat/paper266/ (accessed on 15 March 2010).
- Sollich, P.; Krogh, A. Learning with ensembles: How overfitting can be useful. In Advances in Neural Information Processing Systems; NIPS: Denver, CO, USA, 1995; pp. 190–196. [Google Scholar]
- Shi, L.; Reid, L.H.; Jones, W.D.; Shippy, R.; Warrington, J.A.; Baker, S.C.; Collins, P.J.; De Longueville, F.; Kawasaki, E.S.; Lee, K.Y.; et al. The MicroArray Quality Control (MAQC) project shows inter-and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol. 2006, 24, 1151. [Google Scholar] [PubMed] [Green Version]
- Chen, J.J.; Hsueh, H.M.; Delongchamp, R.R.; Lin, C.J.; Tsai, C.A. Reproducibility of microarray data: A further analysis of microarray quality control (MAQC) data. BMC Bioinform. 2007, 8, 412. [Google Scholar] [CrossRef] [Green Version]
- Guilleaume, B. Microarray Quality Control. By Wei Zhang, Ilya Shmulevich and Jaakko Astola. Proteomics 2005, 5, 4638–4639. [Google Scholar] [CrossRef]
- Chicco, D.; Jurman, G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genom. 2020, 21, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Z.; Łabaj, P.P.; Li, S.; Thierry-Mieg, J.; Thierry-Mieg, D.; Shi, W.; Wang, C.; Schroth, G.P.; Setterquist, R.A.; Thompson, J.F.; et al. SEQC/MAQC-III Consortium: A comprehensive assessment of 521 RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control 522 Consortium. Nat. Biotechnol. 2014, 32, 903–914. [Google Scholar]
- Nguyen, X.V.; Chan, J.; Romano, S.; Bailey, J. Effective global approaches for mutual information based feature selection. In Proceedings of the 20th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, New York, NY, USA, 24–27 August 2014; ACM: New York, NY, USA, 2014; pp. 512–521. [Google Scholar]
- Potharaju, S.P.; Sreedevi, M. Distributed feature selection (DFS) strategy for microarray gene expression data to improve the classification performance. Clin. Epidemiol. Glob. Health 2019, 7, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Palade, V.; Xu, Y. Neuro-fuzzy ensemble approach for microarray cancer gene expression data analysis. In Proceedings of the 2006 International Symposium on Evolving Fuzzy Systems, Ambleside, UK, 7–9 September 2006; pp. 241–246. [Google Scholar]
- Chen, W.; Lu, H.; Wang, M.; Fang, C. Gene expression data classification using artificial neural network ensembles based on samples filtering. In Proceedings of the 2009 International Conference on Artificial Intelligence and Computational Intelligence, Shanghai, China, 7–8 November 2009; Volume 1, pp. 626–628. [Google Scholar]
- Bosio, M.; Salembier, P.; Bellot, P.; Oliveras-Verges, A. Hierarchical clustering combining numerical and biological similarities for gene expression data classification. In Proceedings of the Engineering in Medicine and Biology Society (EMBC), 2013 35th Annual International Conference of the IEEE, Osaka, Japan, 3–7 July 2013; pp. 584–587. [Google Scholar]
- Gashler, M.; Giraud-Carrier, C.; Martinez, T. Decision tree ensemble: Small heterogeneous is better than large homogeneous. In Proceedings of the 2008 Seventh International Conference on Machine Learning and Applications, San Diego, CA, USA, 11–13 December 2008; pp. 900–905. [Google Scholar]
- Wu, Y. Multi-Label Super Learner: Multi-Label Classification and Improving Its Performance Using Heterogenous Ensemble Methods; Wellesley College: Wellesley, MA, USA, 2018. [Google Scholar]
- Yu, Y.; Wang, Y.; Furst, J.; Raicu, D. Identifying Diagnostically Complex Cases Through Ensemble Learning. In International Conference on Image Analysis and Recognition (ICIAR); Lecture Notes in Computer Science, Volume 11663; Springer: Cham Switzerland, 2019; pp. 316–324. [Google Scholar]
- Ayadi, W.; Elloumi, M. Biclustering of microarray data. In Algorithms in Computational Molecular Biology: Techniques, Approaches and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2011; pp. 651–663. [Google Scholar]
- Mohapatra, P.; Chakravarty, S.; Dash, P. Microarray medical data classification using kernel ridge regression and modified cat swarm optimization based gene selection system. Swarm Evol. Comput. 2016, 28, 144–160. [Google Scholar] [CrossRef]
- Ravishankar, H.; Sudhakar, P.; Venkataramani, R.; Thiruvenkadam, S.; Annangi, P.; Babu, N.; Vaidya, V. Understanding the mechanisms of deep transfer learning for medical images. arXiv 2017, arXiv:1704.06040. [Google Scholar]
- Polat, K.; Güneş, S. A novel hybrid intelligent method based on C4. 5 decision tree classifier and one-against-all approach for multi-class classification problems. Expert Syst. Appl. 2009, 36, 1587–1592. [Google Scholar] [CrossRef]
- Friedman, N.; Linial, M.; Nachman, I.; Pe’er, D. Using Bayesian networks to analyze expression data. J. Comput. Biol. 2000, 7, 601–620. [Google Scholar] [CrossRef] [PubMed]
- Hastie, T.; Rosset, S.; Zhu, J.; Zou, H. Multi-class adaboost. Stat. Interface 2009, 2, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Kégl, B. The return of AdaBoost. MH: Multi-class Hamming trees. arXiv 2013, arXiv:1312.6086. [Google Scholar]
- Geurts, P.; Ernst, D.; Wehenkel, L. Extremely randomized trees. Mach. Learn. 2006, 63, 3–42. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Wang, L. Dimensionality dependent PAC-Bayes margin bound. In Advances in Neural Information Processing Systems; Curran Associates, Inc.: New York, NY, USA, 2012; pp. 1034–1042. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Brown, M.P.; Grundy, W.N.; Lin, D.; Cristianini, N.; Sugnet, C.W.; Furey, T.S.; Ares, M.; Haussler, D. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc. Natl. Acad. Sci. USA 2000, 97, 262–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Gong, D.W.; Cheng, J. Multi-objective particle swarm optimization approach for cost-based feature selection in classification. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 14, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Annavarapu, C.S.R.; Dara, S.; Banka, H. Cancer microarray data feature selection using multi-objective binary particle swarm optimization algorithm. EXCLI J. 2016, 15, 460. [Google Scholar]
- Plagianakos, V.; Tasoulis, D.; Vrahatis, M. Gene Expression Data Classification Using Computational Intelligence Techniques. 2005. Available online: https://thalis.math.upatras.gr/~dtas/papers/PlagianakosTV2005b.pdf (accessed on 15 March 2005).
- Bosio, M.; Bellot, P.; Salembier, P.; Verge, A.O. Ensemble learning and hierarchical data representation for microarray classification. In Proceedings of the 13th IEEE International Conference on BioInformatics and BioEngineering, Chania, Greece, 10–13 November 2013; pp. 1–4. [Google Scholar]
- Luo, J.; Schumacher, M.; Scherer, A.; Sanoudou, D.; Megherbi, D.; Davison, T.; Shi, T.; Tong, W.; Shi, L.; Hong, H.; et al. A comparison of batch effect removal methods for enhancement of prediction performance using MAQC-II microarray gene expression data. Pharmacogenomics J. 2010, 10, 278–291. [Google Scholar] [CrossRef] [Green Version]
- Bosio, M.; Bellot, P.; Salembier, P.; Oliveras-Verges, A. Gene expression data classification combining hierarchical representation and efficient feature selection. J. Biol. Syst. 2012, 20, 349–375. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Endpoint | Accession Code | Features | Training Samples | Validation Samples | ||||
|---|---|---|---|---|---|---|---|---|---|
| +ve | −ve | Total | +ve | −ve | Total | ||||
| Dataset A | Hamner | GSE24061 | 1004004 | 26 | 44 | 70 | 28 | 60 | 88 |
| Dataset B | Iconix | GSE24417 | 0010560 | 73 | 143 | 216 | 57 | 144 | 201 |
| Dataset C | NIEHS | GSE24363 | 0695556 | 79 | 135 | 214 | 78 | 126 | 204 |
| Dataset | ||
|---|---|---|
| A | 0.639793072 | 0.4923244337 |
| B | 0.1025047719 | 0.09680510363 |
| C | 0.6312148229 | 0.6648090879 |
| LIG(i) | |||||
|---|---|---|---|---|---|
| Dataset | |LIG| | Accuracy | MCC | Accuracy | MCC |
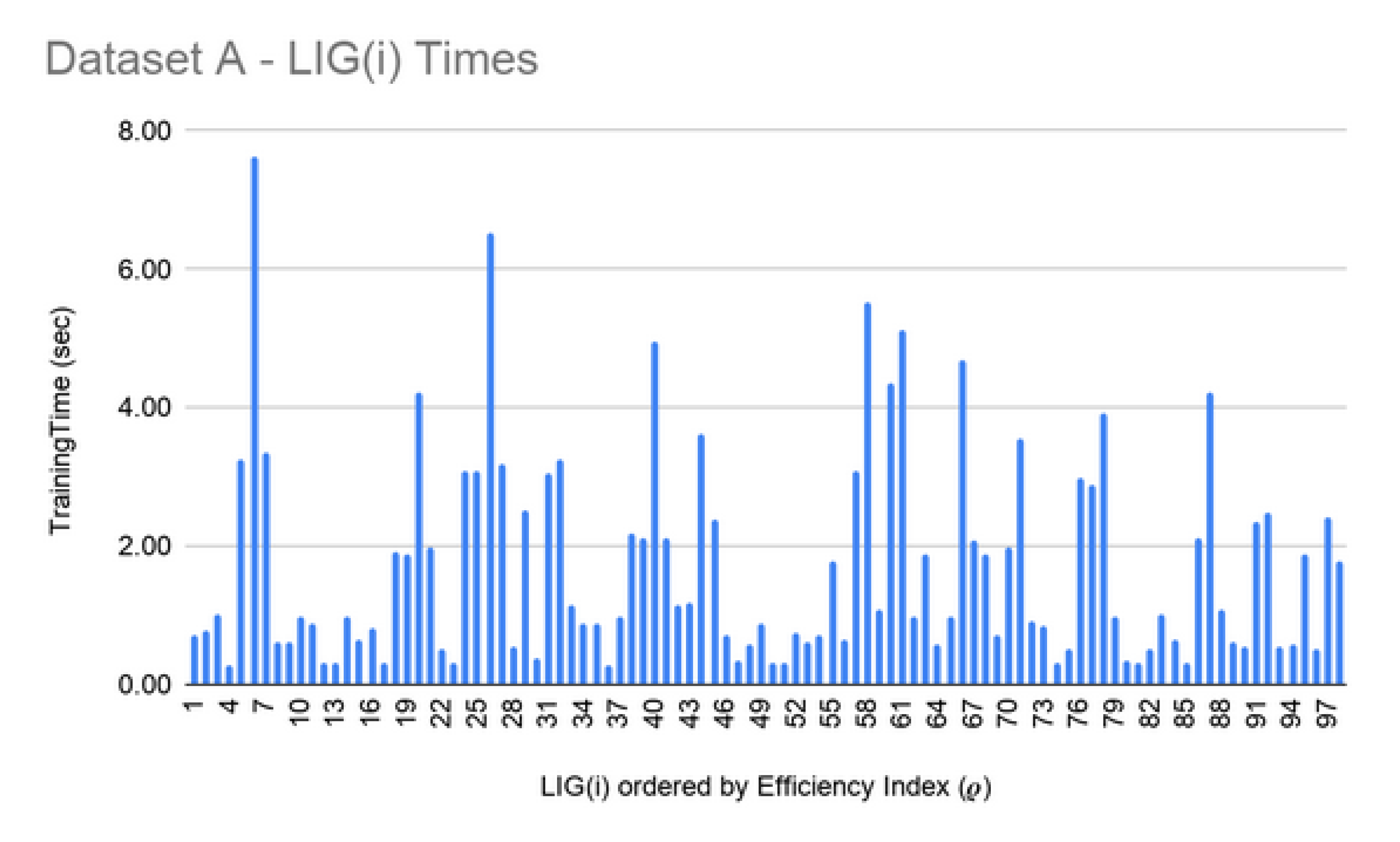
| A | 98 | 75% | 0.89 | 95% | 0.90 |
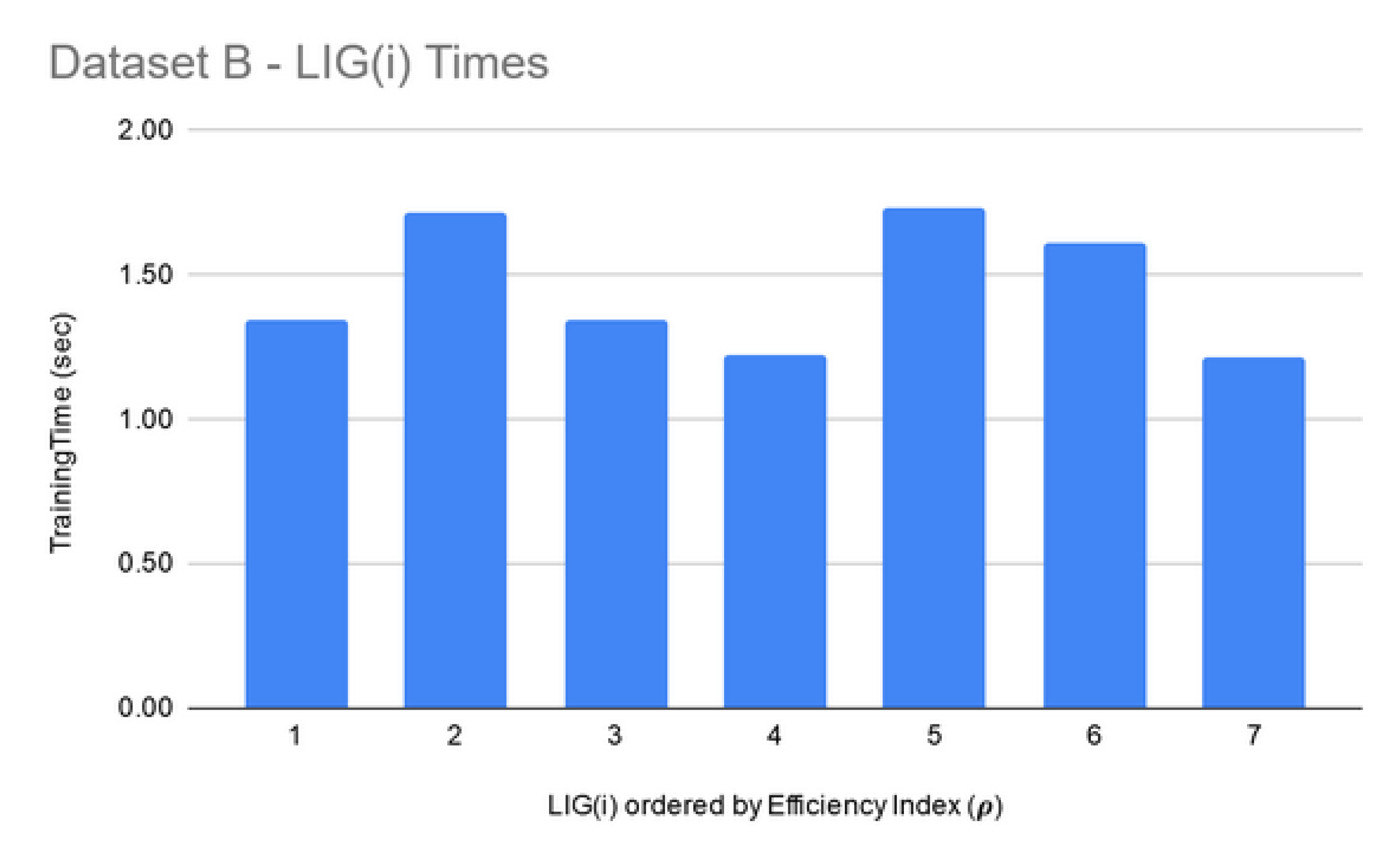
| B | 07 | 69% | 0.18 | 72% | 0.21 |
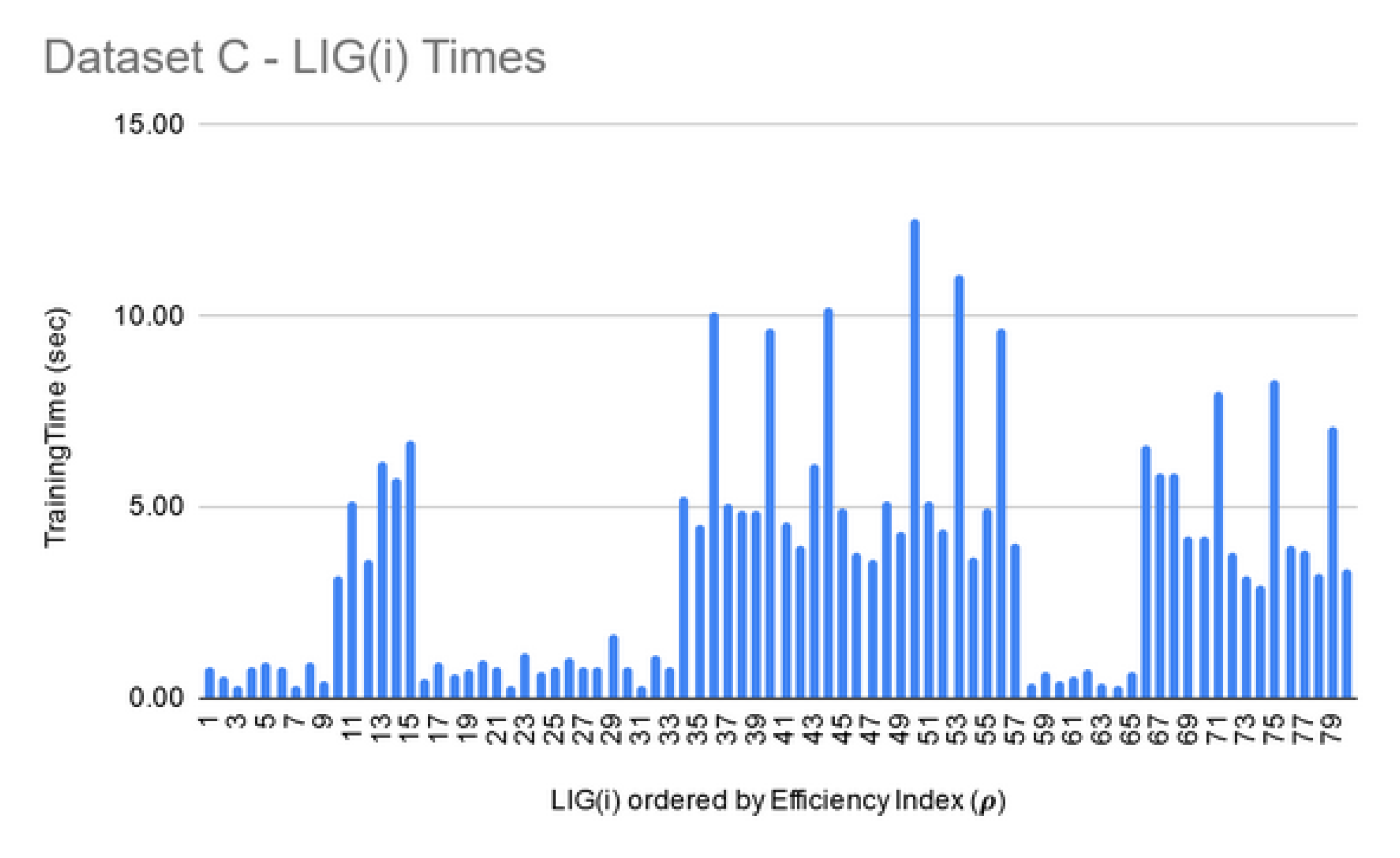
| C | 80 | 88% | 0.76 | 90% | 0.82 |
| FT(i) | |||||
|---|---|---|---|---|---|
| Dataset | |FT| | Accuracy | MCC | Accuracy | MCC |
| A | 199 | 70% | 0.82 | 97% | 0.92 |
| B | 12 | 68% | 0.16 | 72% | 0.08 |
| C | 108 | 90% | 0.81 | 91% | 0.84 |
| Dataset | Measure | Ensembles | |||||
|---|---|---|---|---|---|---|---|
| LIG(i) | FT(i) | LIG | FT | LIG & FT Combined | ITO Tuned | ||
| (X1) | (X2) | (X3) | (X4) | (X5) | (X6) | ||
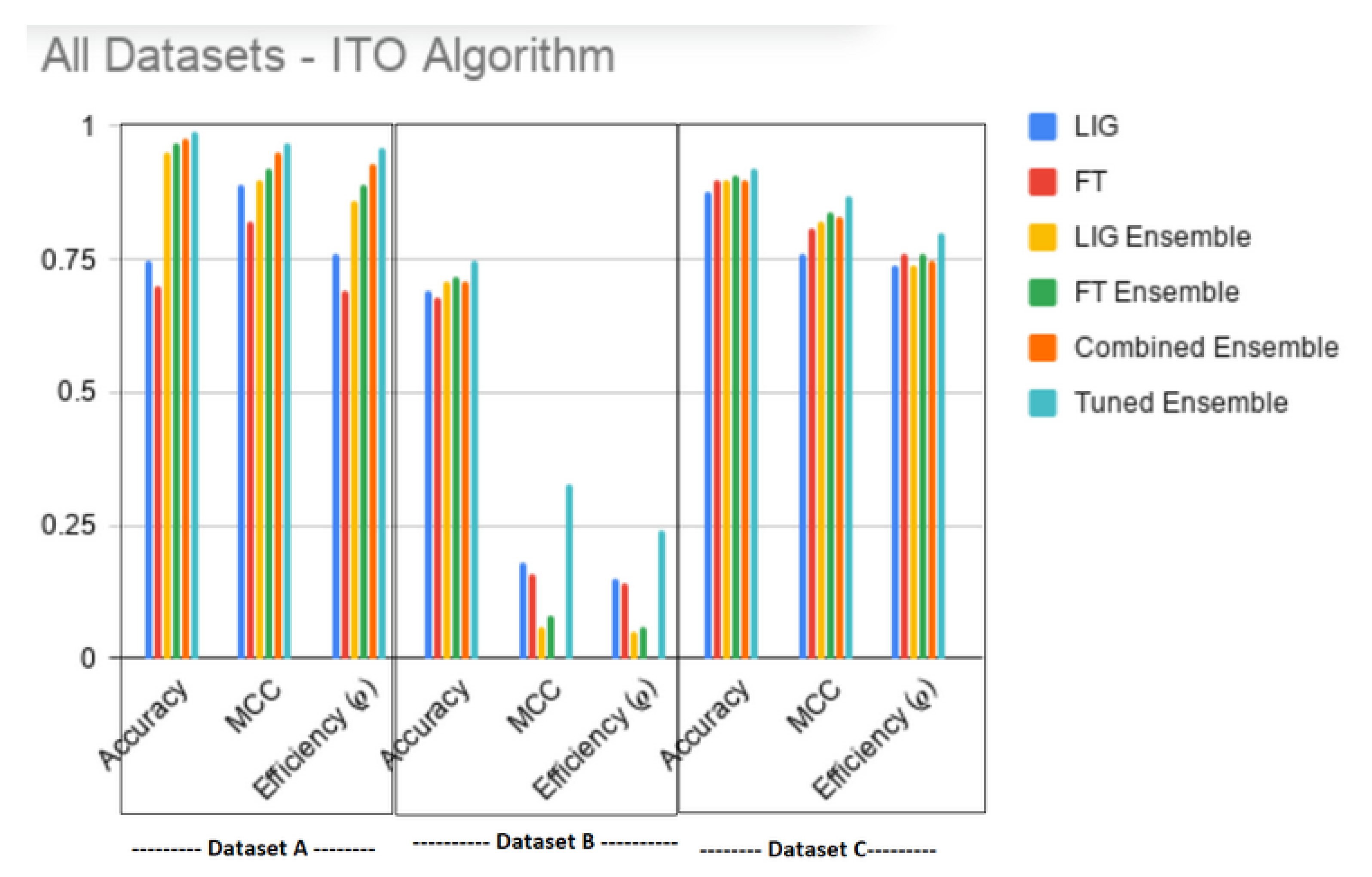
| A | Accuracy | 0.75 | 0.70 | 0.95 | 0.97 | 0.98 | 0.99 |
| MCC | 0.89 | 0.82 | 0.90 | 0.92 | 0.95 | 0.97 | |
| Efficiency () | 0.76 | 0.69 | 0.86 | 0.89 | 0.93 | 0.96 | |
| B | Accuracy | 0.69 | 0.68 | 0.71 | 0.72 | 0.71 | 0.75 |
| MCC | 0.18 | 0.16 | 0.06 | 0.08 | 0.00 | 0.33 | |
| Efficiency () | 0.15 | 0.14 | 0.05 | 0.06 | 0.00 | 0.24 | |
| C | Accuracy | 0.88 | 0.90 | 0.90 | 0.91 | 0.90 | 0.92 |
| MCC | 0.76 | 0.81 | 0.82 | 0.84 | 0.83 | 0.87 | |
| Efficiency () | 0.74 | 0.76 | 0.74 | 0.76 | 0.75 | 0.80 | |
| Dataset | Measure | ITO Improvement % Age Over | ||||
|---|---|---|---|---|---|---|
| LIG | FT | LIG Ensemble | FT Ensemble | LIG & FT Combined Ensemble | ||
| (X6−X1)/X6% | (X6−X2)/X6% | (X6−X3)/X6% | (X6−X4)/X6% | (X6−X5)/X6% | ||
| A | Accuracy | 24.24% | 29.29% | 04.04% | 02.02% | 01.01% |
| MCC | 08.25% | 15.46% | 07.22% | 05.15% | 02.06% | |
| Efficiency () | 20.83% | 28.12% | 10.42% | 07.29% | 03.12% | |
| B | Accuracy | 08.00% | 09.33% | 05.33% | 04.00% | 05.33% |
| MCC | 45.45% | 51.52% | 81.82% | 75.76% | 100% | |
| Efficiency () | 37.5 0% | 41.66% | 79.17% | 75.00% | 100% | |
| C | Accuracy | 04.35% | 02.17% | 02.17% | 01.07% | 02.17% |
| MCC | 12.64% | 06.90% | 05.75% | 03.45% | 04.60% | |
| Efficiency () | 07.50% | 05.00% | 07.50% | 05.00% | 06.25% | |
| Method | MCC | Accuracy |
|---|---|---|
| MAQC-II [12] | 0.210 | - |
| AID [56] | 0.293 | - |
| Kun [56] | 0.407 | - |
| Kun [56] | 0.303 | - |
| Kun [56] | 0.346 | - |
| EJLR [57] | 0.57 | - |
| Monte Carlo simulation as reported in [36] | 0.270 | 67.3% |
| ITO algorithm | 0.950 | 98% |
| Method | MCC | Accuracy |
|---|---|---|
| MAQC-II [12] | 0.830 | - |
| AID [56] | 0.793 | - |
| Kun [56] | 0.812 | - |
| Kun [56] | 0.804 | - |
| Kun [56] | 0.781 | - |
| Kun [56] | 0.792 | - |
| t-test with KNN (Mean Centering) [57] | 0.80 | - |
| Monte Carlo simulation as reported in [58] | 0.795 | 90.25% |
| ITO algorithm | 0.870 | 92% |
| Method | MCC | Accuracy |
|---|---|---|
| MAQC-II [12] | 0.42 | - |
| Ratio-G [57] | 0.50 | - |
| ITO algorithm | 0.33 | 75% |
| Data Set | Number of Features | Min Time for FSS (Size 10) | Max Time for FSS (Size 250) |
|---|---|---|---|
| A | 1,004,004 | 26 h | >72 h |
| B | 10,560 | 50 min | 2.6 h |
| C | 695,556 | 24 h | 72 h |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zahoor, J.; Zafar, K. Classification of Microarray Gene Expression Data Using an Infiltration Tactics Optimization (ITO) Algorithm. Genes 2020, 11, 819. https://doi.org/10.3390/genes11070819
Zahoor J, Zafar K. Classification of Microarray Gene Expression Data Using an Infiltration Tactics Optimization (ITO) Algorithm. Genes. 2020; 11(7):819. https://doi.org/10.3390/genes11070819
Chicago/Turabian StyleZahoor, Javed, and Kashif Zafar. 2020. "Classification of Microarray Gene Expression Data Using an Infiltration Tactics Optimization (ITO) Algorithm" Genes 11, no. 7: 819. https://doi.org/10.3390/genes11070819
APA StyleZahoor, J., & Zafar, K. (2020). Classification of Microarray Gene Expression Data Using an Infiltration Tactics Optimization (ITO) Algorithm. Genes, 11(7), 819. https://doi.org/10.3390/genes11070819