RNA-Seq Analysis Reveals Localization-Associated Alternative Splicing across 13 Cell Lines


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. RNA-Seq Data and Bioinformatics
2.2. and
2.3. Gene Ontology (GO) Enrichment Analysis
2.4. Nonsense-Mediated RNA Decay (NMD) Sensitivity
2.5. RNA Structure Context Analysis
2.6. RBP-Binding Prediction and Repeat Sequence Analysis
3. Results
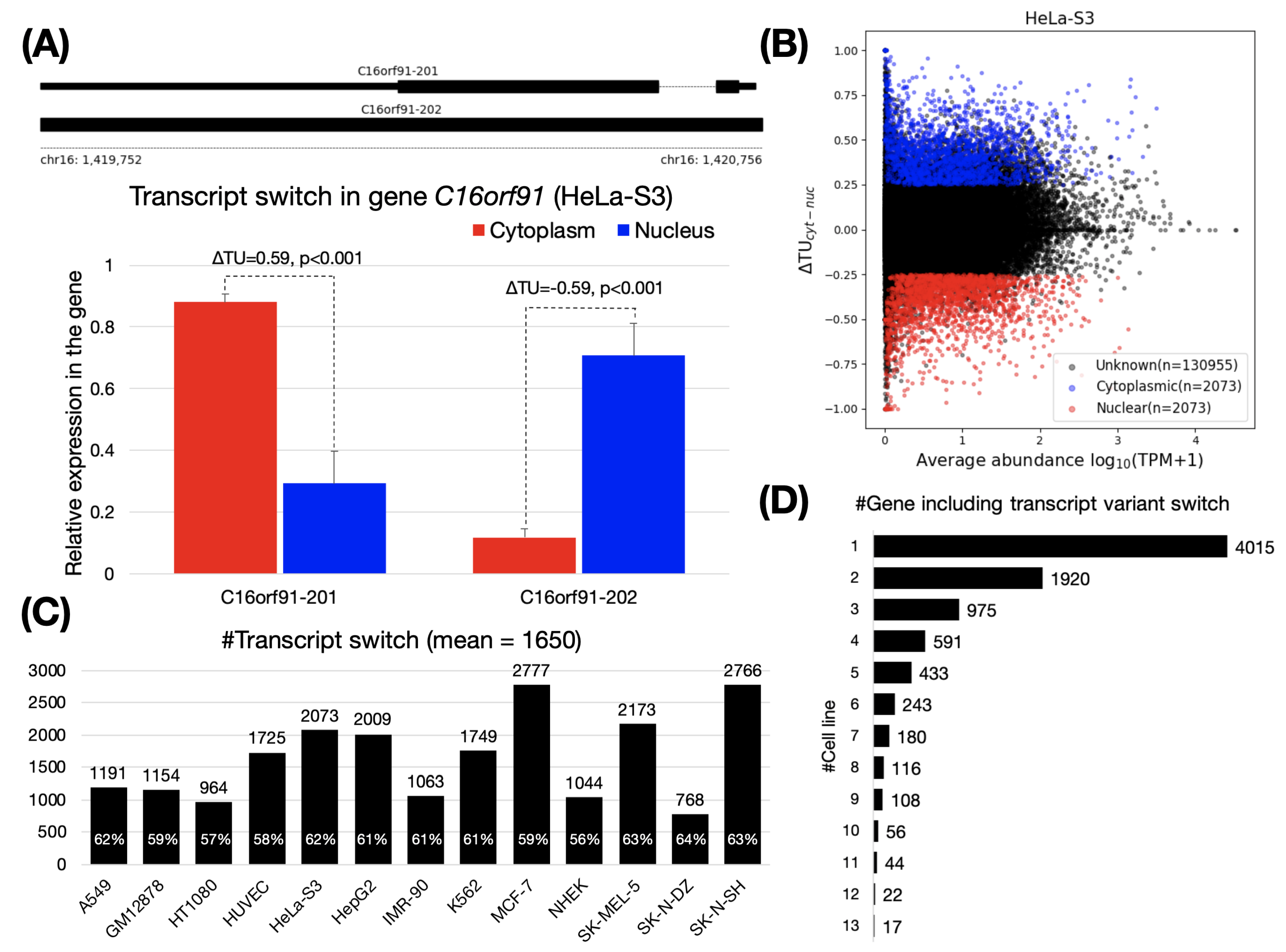
3.1. Thousands of Transcript Switches between Cytoplasm and Nucleus Were Identified across Thirteen Human Cell Lines
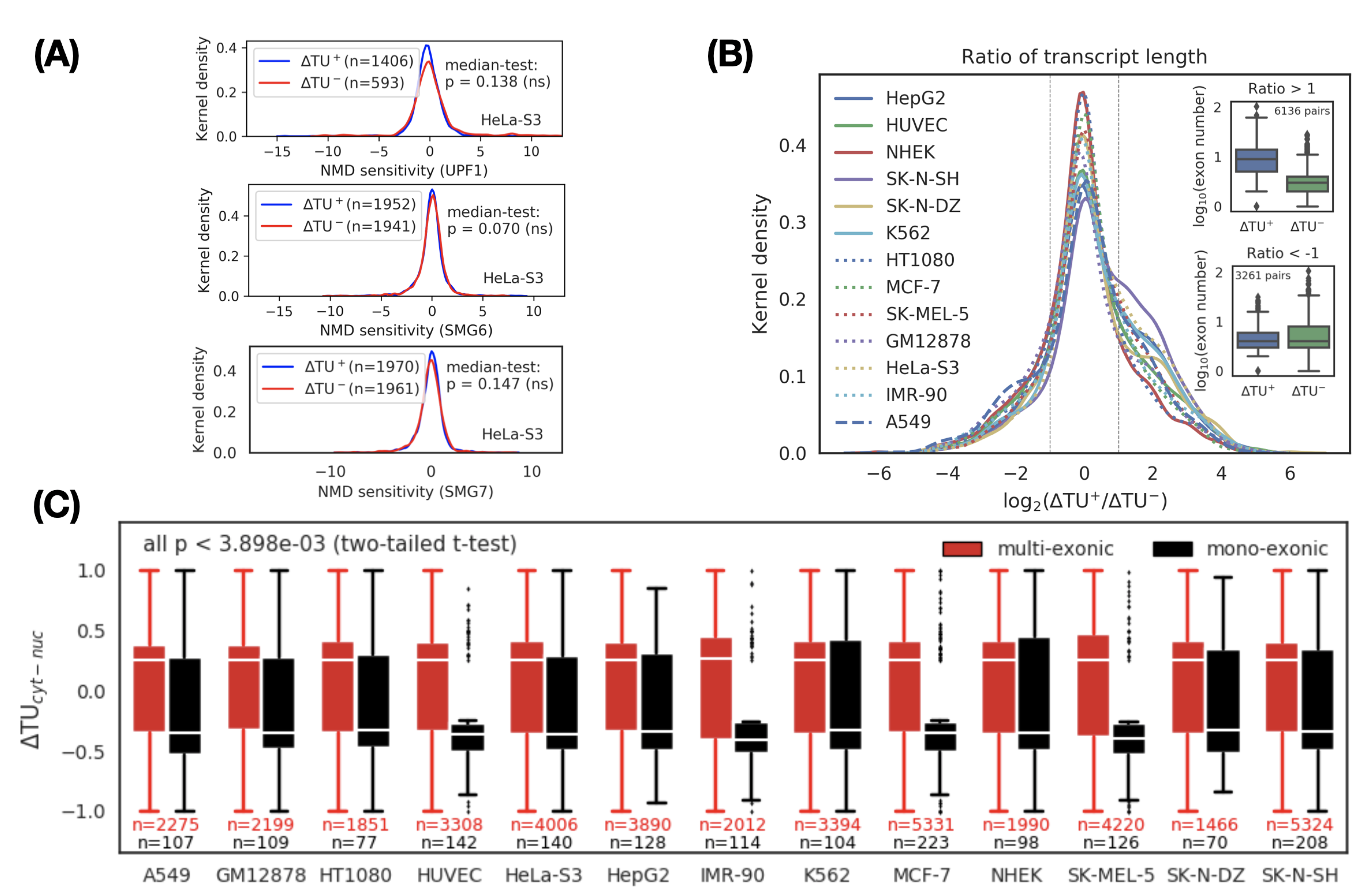
3.2. A Transcript Switch Is Associated with RNA Splicing Rather Than RNA Degradation
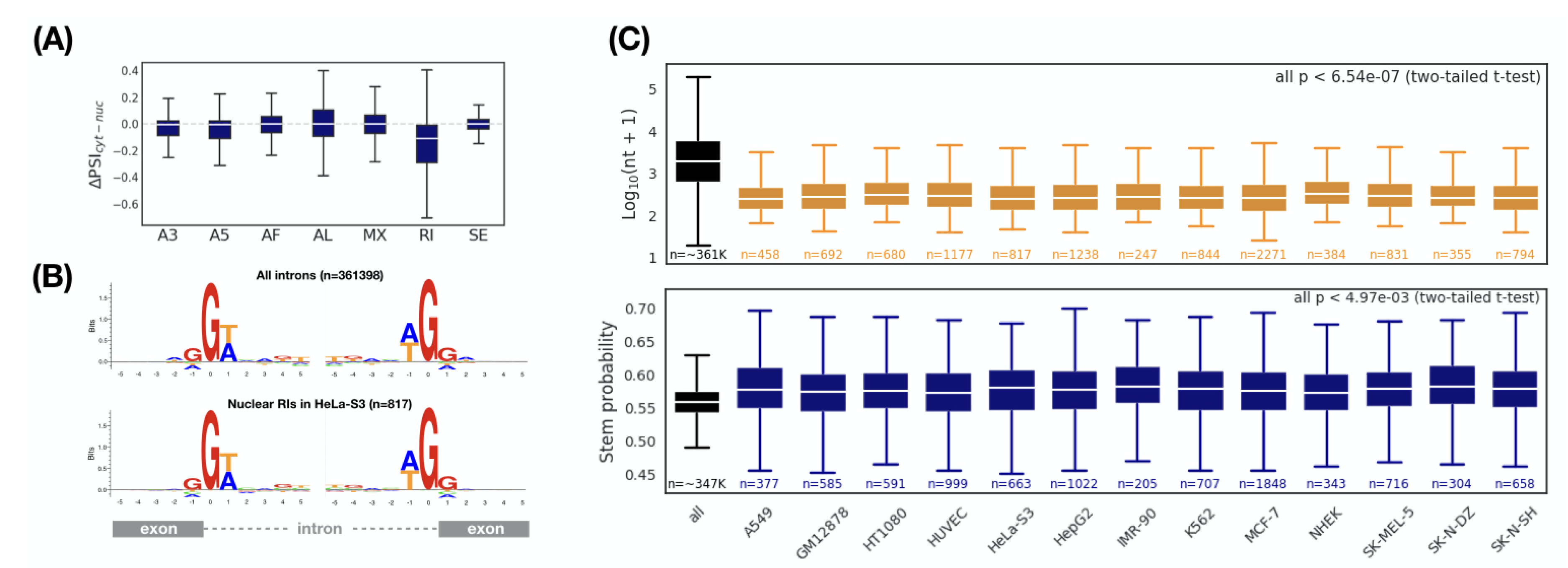
3.3. Enrichment and Characterization of Retained Introns in the Nuclear Transcripts
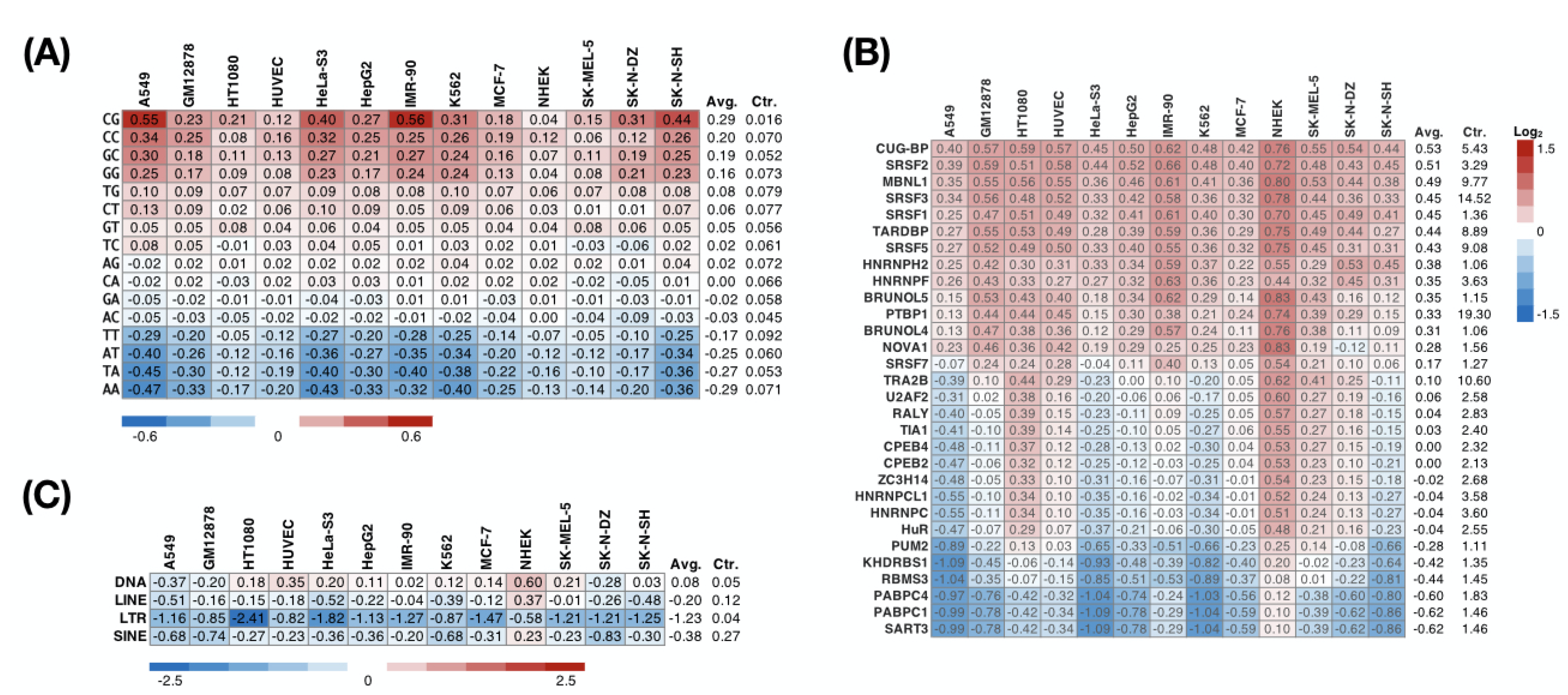
3.4. The Preferences of RNA-Binding Proteins on Retained Introns Suggest Their Role in Nuclear Localization
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RNA FISH | RNA fluorescent in situ hybridization |
| RNA-seq | RNA sequencing |
| RBP | RNA-binding protein |
| NLS | nuclear signal sequence |
| change of transcript usage | |
| GO | Gene Ontology |
| MF | molecular function |
| BP | biological processes |
| CC | and cellular component |
| NMD | nonsense-mediated decay |
| change of splicing inclusion | |
| TE | transposon element |
| RI | retained intron |
| SINE | Short interspersed nuclear element |
| LINE | Long interspersed nuclear element |
| LTR | Long terminal repeat |
| GCR | genetic compensation response |
| EJC | exon-exon junction complex |
| TPM | transcripts per million |
| A3 | alternative 3 splice-site |
| A5 | alternative 5 splice-site |
| AF | alternative first exon |
| AL | alternative last exon |
| MX | mutually exclusive exon |
| SE | skipping exon |
References
- Brett, D.; Pospisil, H.; Valcárcel, J.; Reich, J.; Bork, P. Alternative splicing and genome complexity. Nat. Genet. 2002, 30, 29–30. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Graveley, B.R. Alternative splicing: Increasing diversity in the proteomic world. Trends Genet. 2001, 17, 100–107. [Google Scholar] [CrossRef]
- Stamm, S.; Ben-Ari, S.; Rafalska, I.; Tang, Y.; Zhang, Z.; Toiber, D.; Thanaraj, T.; Soreq, H. Function of alternative splicing. Gene 2005, 344, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cieply, B.; Carstens, R.P. Functional roles of alternative splicing factors in human disease. Wiley Interdiscip. Rev. RNA 2015, 6, 311–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, C.; Zhang, Q.; Chen, Y.; Wang, Y.; Zhang, X.; Liu, Y.; Sun, C.; Zhang, H.; Hoheisel, J.D. Function, clinical application, and strategies of Pre-mRNA splicing in cancer. Cell Death Differ. 2019, 26, 1181–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ule, J.; Blencowe, B.J. Alternative Splicing Regulatory Networks: Functions, Mechanisms, and Evolution. Mol. Cell 2019, 76, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, A.R.; Haimovich, G.; Singer, R.H. In the right place at the right time: Visualizing and understanding mRNA localization. Nat. Rev. Mol. Cell Biol. 2015, 16, 95–109. [Google Scholar] [CrossRef]
- Fasken, M.B.; Corbett, A.H. Mechanisms of nuclear mRNA quality control. RNA Biol. 2009, 6, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Nevo-Dinur, K.; Govindarajan, S.; Amster-Choder, O. Subcellular localization of RNA and proteins in prokaryotes. Trends Genet. 2012, 28, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Dermit, M.; Dodel, M.; Lee, F.C.; Azman, M.S.; Schwenzer, H.; Jones, J.L.; Blagden, S.P.; Ule, J.; Mardakheh, F.K. Subcellular mRNA localization regulates ribosome biogenesis in migrating cells. bioRxiv 2019. [Google Scholar] [CrossRef]
- Bashirullah, A.; Cooperstock, R.L.; Lipshitz, H.D. RNA localization in development. Annu. Rev. Biochem. 1998, 67, 335–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; King, M.L. Sending RNAs into the future: RNA localization and germ cell fate. IUBMB Life 2004, 56, 19–27. [Google Scholar]
- Lawrence, J.B.; Singer, R.H. Intracellular localization of messenger RNAs for cytoskeletal proteins. Cell 1986, 45, 407–415. [Google Scholar] [CrossRef]
- Femino, A.M.; Fay, F.S.; Fogarty, K.; Singer, R.H. Visualization of single RNA transcripts in situ. Science 1998, 280, 585–590. [Google Scholar] [CrossRef] [Green Version]
- Raj, A.; Van Den Bogaard, P.; Rifkin, S.A.; Van Oudenaarden, A.; Tyagi, S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 2008, 5, 877–879. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Daugharthy, E.R.; Scheiman, J.; Kalhor, R.; Yang, J.L.; Ferrante, T.C.; Terry, R.; Jeanty, S.S.F.; Li, C.; Amamoto, R.; et al. Highly multiplexed subcellular RNA sequencing in situ. Science 2014, 343, 1360–1363. [Google Scholar] [CrossRef] [Green Version]
- Taliaferro, J.M.; Wang, E.T.; Burge, C.B. Genomic analysis of RNA localization. RNA Biol. 2014, 11, 1040–1050. [Google Scholar] [CrossRef] [Green Version]
- Fazal, F.M.; Han, S.; Parker, K.R.; Kaewsapsak, P.; Xu, J.; Boettiger, A.N.; Chang, H.Y.; Ting, A.Y. Atlas of subcellular RNA localization revealed by APEX-seq. Cell 2019, 178, 473–490. [Google Scholar] [CrossRef]
- Padron, A.; Iwasaki, S.; Ingolia, N.T. Proximity RNA labeling by APEX-seq reveals the organization of translation initiation complexes and repressive RNA granules. Mol. Cell 2019, 75, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Tan, P.; Wang, L.; Jin, N.; Li, Y.; Zhang, L.; Yang, H.; Hu, Z.; Zhang, L.; Hu, C.; et al. RNALocate: A resource for RNA subcellular localizations. Nucleic Acids Res. 2016, 45, D135–D138. [Google Scholar] [PubMed]
- Mas-Ponte, D.; Carlevaro-Fita, J.; Palumbo, E.; Pulido, T.H.; Guigo, R.; Johnson, R. LncATLAS database for subcellular localization of long noncoding RNAs. RNA 2017, 23, 1080–1087. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Liu, Z.; Wu, J.; Liu, J.h.; Hyder, S.M.; Antoniou, E.; Lubahn, D.B. Identification and characterization of two novel splicing isoforms of human estrogen-related receptor β. J. Clin. Endocrinol. Metab. 2006, 91, 569–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakre, S.; Tussie-Luna, M.I.; Ashworth, T.; Novina, C.D.; Settleman, J.; Sharp, P.A.; Roy, A.L. Opposing functions of TFII-I spliced isoforms in growth factor-induced gene expression. Mol. Cell 2006, 24, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Wiesenthal, A.; Hoffmeister, M.; Siddique, M.; Kovacevic, I.; Oess, S.; Müller-Esterl, W.; Siehoff-Icking, A. NOSTRINβ—A shortened NOSTRIN variant with a role in transcriptional regulation. Traffic 2009, 10, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Bombail, V.; Collins, F.; Brown, P.; Saunders, P.T. Modulation of ERα transcriptional activity by the orphan nuclear receptor ERRβ and evidence for differential effects of long-and short-form splice variants. Mol. Cell. Endocrinol. 2010, 314, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Ferreira, A.M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trincado, J.L.; Entizne, J.C.; Hysenaj, G.; Singh, B.; Skalic, M.; Elliott, D.J.; Eyras, E. SUPPA2: Fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018, 19, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, S.; Park, J.W.; Lu, Z.x.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [Green Version]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g: Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [Green Version]
- Colombo, M.; Karousis, E.D.; Bourquin, J.; Bruggmann, R.; Mühlemann, O. Transcriptome-wide identification of NMD-targeted human mRNAs reveals extensive redundancy between SMG6- and SMG7-mediated degradation pathways. RNA 2017, 23, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukunaga, T.; Ozaki, H.; Terai, G.; Asai, K.; Iwasaki, W.; Kiryu, H. CapR: Revealing structural specificities of RNA-binding protein target recognition using CLIP-seq data. Genome Biol. 2014, 15, R16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz, I.; Kosti, I.; Ares Jr, M.; Cline, M.; Mandel-Gutfreund, Y. RBPmap: A web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 2014, 42, W361–W367. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.; Hubley, R.; Green, P. RepeatMasker Open-4.0, 2013–2015. 2013. Available online: http://www.repeatmasker.org (accessed on 1 October 2019).
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47. [Google Scholar] [CrossRef] [PubMed]
- Marchese, F.P.; Raimondi, I.; Huarte, M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017, 18, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regulapati, R.; Bhasi, A.; Singh, C.K.; Senapathy, P. Origination of the split structure of spliceosomal genes from random genetic sequences. PLoS ONE 2008, 3, e3456. [Google Scholar] [CrossRef]
- Alamancos, G.P.; Pagès, A.; Trincado, J.L.; Bellora, N.; Eyras, E. Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA 2015, 21, 1521–1531. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, M.; Kumagai, Y.; Standley, D.M.; Sarai, A.; Mizuguchi, K.; Ahmad, S. Prediction of dinucleotide-specific RNA-binding sites in proteins. BMC Bioinform. 2011, 12, S5. [Google Scholar] [CrossRef] [Green Version]
- Lubelsky, Y.; Ulitsky, I. Sequences enriched in Alu repeats drive nuclear localization of long RNAs in human cells. Nature 2018, 555, 107–111. [Google Scholar] [CrossRef]
- Park, E.; Pan, Z.; Zhang, Z.; Lin, L.; Xing, Y. The expanding landscape of alternative splicing variation in human populations. Am. J. Hum. Genet. 2018, 102, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Wen, W.X.; Mead, A.J.; Thongjuea, S. Technological advances and computational approaches for alternative splicing analysis in single cells. Comput. Struct. Biotechnol. J. 2020, 18, 332–343. [Google Scholar] [CrossRef]
- Parker, M.T.; Knop, K.; Sherwood, A.V.; Schurch, N.J.; Mackinnon, K.; Gould, P.D.; Hall, A.J.; Barton, G.J.; Simpson, G.G. Nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and m6A modification. eLife 2020, 9, e49658. [Google Scholar] [CrossRef]
- Tang, A.D.; Soulette, C.M.; van Baren, M.J.; Hart, K.; Hrabeta-Robinson, E.; Wu, C.J.; Brooks, A.N. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kloc, M.; Wilk, K.; Vargas, D.; Shirato, Y.; Bilinski, S.; Etkin, L.D. Potential structural role of non-coding and coding RNAs in the organization of the cytoskeleton at the vegetal cortex of Xenopus oocytes. Development 2005, 132, 3445–3457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenny, A.; Hachet, O.; Závorszky, P.; Cyrklaff, A.; Weston, M.D.; St Johnston, D.; Erdélyi, M.; Ephrussi, A. A translation-independent role of oskar RNA in early Drosophila oogenesis. Development 2006, 133, 2827–2833. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Kumari, P.; Gilligan, P.; Quach, H.N.B.; Mathavan, S.; Sampath, K. Dorsal activity of maternal squint is mediated by a non-coding function of the RNA. Development 2012, 139, 2903–2915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shevtsov, S.P.; Dundr, M. Nucleation of nuclear bodies by RNA. Nat. Cell Biol. 2011, 13, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Jansen, G.; Groenen, P.J.; Bächner, D.; Jap, P.H.; Coerwinkel, M.; Oerlemans, F.; van den Broek, W.; Gohlsch, B.; Pette, D.; Plomp, J.J.; et al. Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat. Genet. 1996, 13, 316. [Google Scholar] [CrossRef]
- Gong, C.; Maquat, L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 2011, 470, 284–288. [Google Scholar] [CrossRef] [Green Version]
- Chooniedass-Kothari, S.; Emberley, E.; Hamedani, M.; Troup, S.; Wang, X.; Czosnek, A.; Hube, F.; Mutawe, M.; Watson, P.; Leygue, E. The steroid receptor RNA activator is the first functional RNA encoding a protein. FEBS Lett. 2004, 566, 43–47. [Google Scholar] [CrossRef]
- Hubé, F.; Velasco, G.; Rollin, J.; Furling, D.; Francastel, C. Steroid receptor RNA activator protein binds to and counteracts SRA RNA-mediated activation of MyoD and muscle differentiation. Nucleic Acids Res. 2010, 39, 513–525. [Google Scholar] [CrossRef] [Green Version]
- Lanz, R.B.; McKenna, N.J.; Onate, S.A.; Albrecht, U.; Wong, J.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell 1999, 97, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A.; Kontarakis, Z.; Gerri, C.; Nolte, H.; Hölper, S.; Krüger, M.; Stainier, D.Y. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 2015, 524, 230. [Google Scholar] [CrossRef] [PubMed]
- El-Brolosy, M.A.; Stainier, D.Y. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017, 13, e1006780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Brolosy, M.A.; Kontarakis, Z.; Rossi, A.; Kuenne, C.; Günther, S.; Fukuda, N.; Kikhi, K.; Boezio, G.L.M.; Takacs, C.M.; Lai, S.L.; et al. Genetic compensation triggered by mutant mRNA degradation. Nature 2019, 568, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhu, P.; Shi, H.; Guo, L.; Zhang, Q.; Chen, Y.; Chen, S.; Zhang, Z.; Peng, J.; Chen, J. PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature 2019, 568, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Le Hir, H.; Gatfield, D.; Izaurralde, E.; Moore, M.J. The exon–exon junction complex provides a binding platform for factors involved in mRNA export and nonsense-mediated mRNA decay. EMBO J. 2001, 20, 4987–4997. [Google Scholar] [CrossRef] [Green Version]
- Reed, R.; Hurt, E. A conserved mRNA export machinery coupled to pre-mRNA splicing. Cell 2002, 108, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Gonzàlez-Porta, M.; Frankish, A.; Rung, J.; Harrow, J.; Brazma, A. Transcriptome analysis of human tissues and cell lines reveals one dominant transcript per gene. Genome Biol. 2013, 14, R70. [Google Scholar] [CrossRef] [Green Version]
- Boutz, P.L.; Bhutkar, A.; Sharp, P.A. Detained introns are a novel, widespread class of post-transcriptionally spliced introns. Genes Dev. 2015, 29, 63–80. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Kertesz, M.; Spitale, R.C.; Segal, E.; Chang, H.Y. Understanding the transcriptome through RNA structure. Nat. Rev. Genet. 2011, 12, 641–655. [Google Scholar] [CrossRef] [Green Version]
- Warf, M.B.; Berglund, J.A. MBNL binds similar RNA structures in the CUG repeats of myotonic dystrophy and its pre-mRNA substrate cardiac troponin T. RNA 2007, 13, 2238–2251. [Google Scholar] [CrossRef] [Green Version]
- Taylor, K.; Sznajder, Ł.J.; Cywoniuk, P.; Thomas, J.D.; Swanson, M.S.; Sobczak, K. MBNL splicing activity depends on RNA binding site structural context. Nucleic Acids Res. 2018, 46, 9119–9133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, M.; Mohan, A.; Batra, R.; Lee, K.Y.; Charizanis, K.; Gómez, F.J.F.; Eddarkaoui, S.; Sergeant, N.; Buée, L.; Kimura, T.; et al. MBNL sequestration by toxic RNAs and RNA misprocessing in the myotonic dystrophy brain. Cell Rep. 2015, 12, 1159–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.Y.; Chang, K.T.; Lin, Y.M.; Kuo, T.Y.; Wang, G.S. Ubiquitination of MBNL1 is required for its cytoplasmic localization and function in promoting neurite outgrowth. Cell Rep. 2018, 22, 2294–2306. [Google Scholar] [CrossRef] [PubMed]
- Valencia, P.; Dias, A.P.; Reed, R. Splicing promotes rapid and efficient mRNA export in mammalian cells. Proc. Natl. Acad. Sci. USA 2008, 105, 3386–3391. [Google Scholar] [CrossRef] [Green Version]
- Zuckerman, B.; Ulitsky, I. Predictive models of subcellular localization of long RNAs. RNA 2019, 25, 557–572. [Google Scholar] [CrossRef]
- Soneson, C.; Yao, Y.; Bratus-Neuenschwander, A.; Patrignani, A.; Robinson, M.D.; Hussain, S. A comprehensive examination of Nanopore native RNA sequencing for characterization of complex transcriptomes. Nat. Commun. 2019, 10, 3359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Workman, R.E.; Tang, A.D.; Tang, P.S.; Jain, M.; Tyson, J.R.; Razaghi, R.; Zuzarte, P.C.; Gilpatrick, T.; Payne, A.; Quick, J.; et al. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat. Methods 2019, 16, 1297–1305. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, C.; Hamada, M. RNA-Seq Analysis Reveals Localization-Associated Alternative Splicing across 13 Cell Lines. Genes 2020, 11, 820. https://doi.org/10.3390/genes11070820
Zeng C, Hamada M. RNA-Seq Analysis Reveals Localization-Associated Alternative Splicing across 13 Cell Lines. Genes. 2020; 11(7):820. https://doi.org/10.3390/genes11070820
Chicago/Turabian StyleZeng, Chao, and Michiaki Hamada. 2020. "RNA-Seq Analysis Reveals Localization-Associated Alternative Splicing across 13 Cell Lines" Genes 11, no. 7: 820. https://doi.org/10.3390/genes11070820
APA StyleZeng, C., & Hamada, M. (2020). RNA-Seq Analysis Reveals Localization-Associated Alternative Splicing across 13 Cell Lines. Genes, 11(7), 820. https://doi.org/10.3390/genes11070820