A Private 16q24.2q24.3 Microduplication in a Boy with Intellectual Disability, Speech Delay and Mild Dysmorphic Features

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genomic DNA Extraction and Quantification
2.2. SNP-Array Analysis
2.3. Real Time Quantitative PCR
3. Results
3.1. Clinical Description
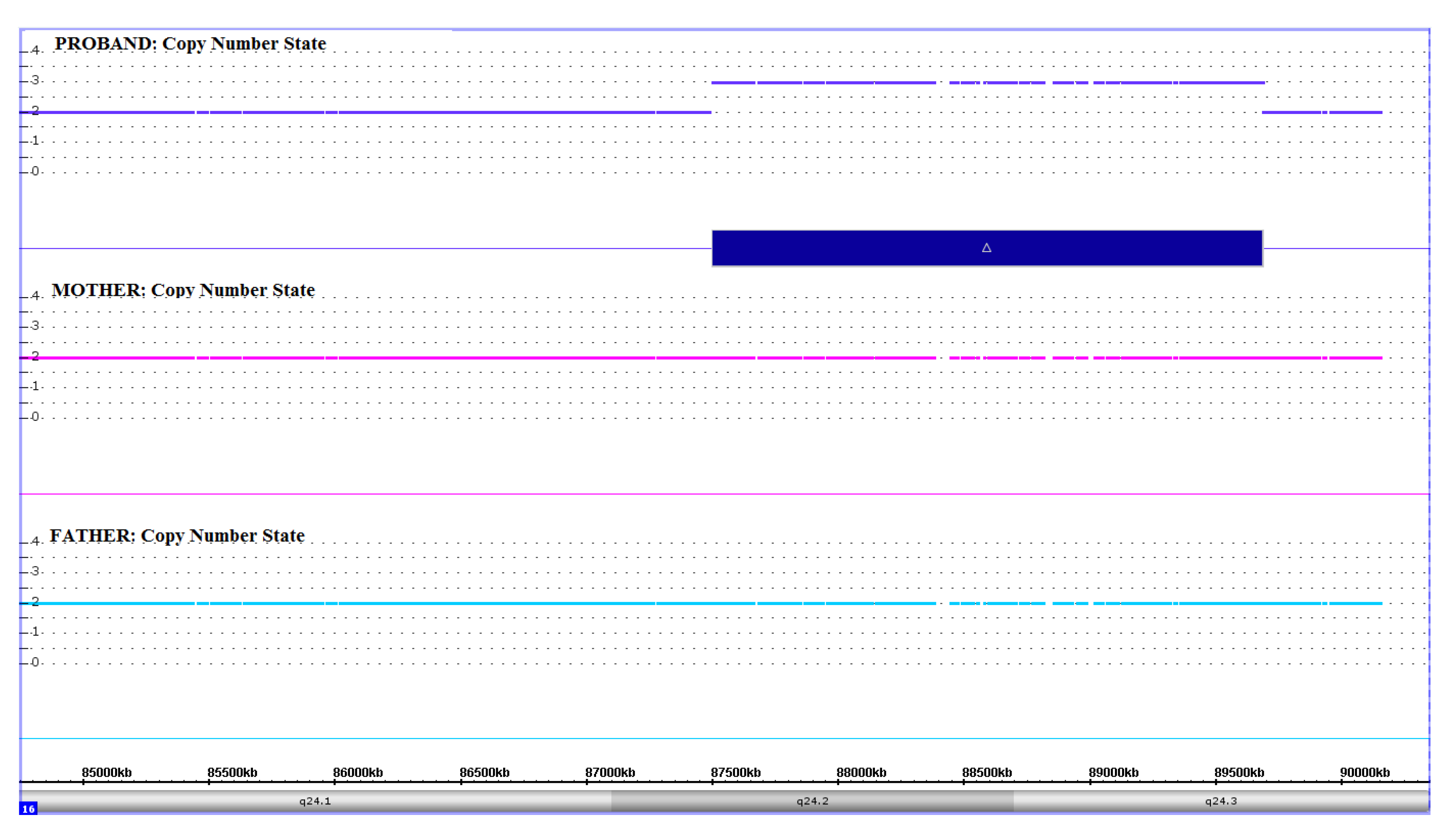
3.2. Molecular Findings
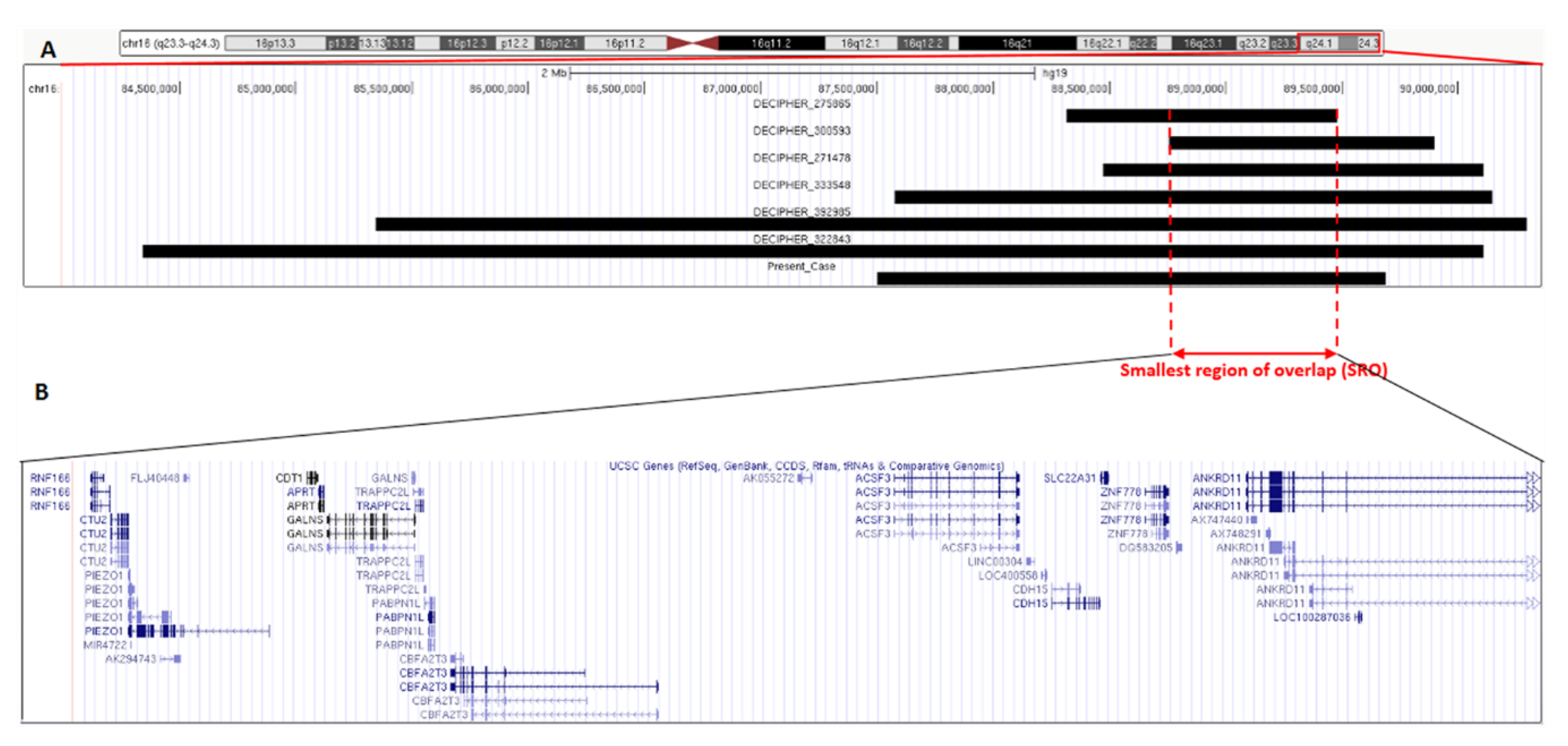
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shaw-Smith, C.; Redon, R.; Rickman, L.; Rio, M.; Willatt, L.; Fiegler, H.; Firth, H.; Sanlaville, D.; Winter, R.; Colleaux, L.; et al. Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J. Med Genet. 2004, 41, 241–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menten, B.; Maas, N.; Thienpont, B.; Buysse, K.; Vandesompele, J.; Melotte, C.; De Ravel, T.; Van Vooren, S.; Balikova, I.; Backx, L.; et al. Emerging patterns of cryptic chromosomal imbalance in patients with idiopathic mental retardation and multiple congenital anomalies: A new series of 140 patients and review of published reports. J. Med Genet. 2006, 43, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, A.; Doccini, V.; Bernardini, L.; Novelli, A.; Loddo, S.; Capalbo, A.; Filippi, T.; Carey, J.C. Confirmation of chromosomal microarray as a first-tier clinical diagnostic test for individuals with developmental delay, intellectual disability, autism spectrum disorders and dysmorphic features. Eur. J. Paediatr. Neurol. 2013, 17, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Slavotinek, A.M. Novel microdeletion syndromes detected by chromosome microarrays. Qual. Life Res. 2008, 124, 1–17. [Google Scholar] [CrossRef]
- Jacobs, H. Don’t ask, don’t tell, don’t publish. EMBO Rep. 2012, 13, 393. [Google Scholar] [CrossRef] [Green Version]
- Nevado, J.; Mergener, R.; Palomares-Bralo, M.; Souza, K.R.; Vallespín, E.; Mena, M.D.R.; Martinez-Glez, V.; Mori, M.Á.; Santos-Simarro, F.; García-Miñaur, S.; et al. New microdeletion and microduplication syndromes: A comprehensive review. Genet. Mol. Boil. 2014, 37, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Watson, C.T.; Marques-Bonet, T.; Sharp, A.J.; Mefford, H.C. The Genetics of Microdeletion and Microduplication Syndromes: An Update. Annu. Rev. Genom. Hum. Genet. 2014, 15, 215–244. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, O.; Fichera, M.; Palumbo, P.; Rizzo, R.; Mazzolla, E.; Cocuzza, D.M.; Carella, M.; Mattina, T. TBR1 is the candidate gene for intellectual disability in patients with a 2q24.2 interstitial deletion. Am. J. Med Genet. Part A 2014, 164, 828–833. [Google Scholar] [CrossRef]
- Kearney, H.M.; Thorland, E.C.; Brown, K.K.; Quintero-Rivera, F.; South, S.T. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet. Med. 2011, 13, 680–685. [Google Scholar] [CrossRef] [Green Version]
- Willemsen, M.; A Fernandez, B.; A Bacino, C.; Gerkes, E.; De Brouwer, A.P.; Pfundt, R.; Sikkema-Raddatz, B.; Scherer, S.W.; Marshall, C.R.; Potocki, L.; et al. Identification of ANKRD11 and ZNF778 as candidate genes for autism and variable cognitive impairment in the novel 16q24.3 microdeletion syndrome. Eur. J. Hum. Genet. 2009, 18, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirmaci, A.; Spiliopoulos, M.; Brancati, F.; Powell, E.; Duman, D.; Abrams, A.; Bademci, G.; Agolini, E.; Guo, S.; Konuk, B.; et al. Mutations in ANKRD11 Cause KBG Syndrome, Characterized by Intellectual Disability, Skeletal Malformations, and Macrodontia. Am. J. Hum. Genet. 2011, 89, 289–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo-Castro, A.; Brancati, F.; Digilio, M.C.; Garaci, F.G.; Bollero, P.; Alfieri, P.; Curatolo, P. Neurobehavioral phenotype observed in KBG syndrome caused byANKRD11mutations. Am. J. Med Genet. Part B: Neuropsychiatr. Genet. 2012, 162, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Crippa, M.; Rusconi, D.; Castronovo, C.; Bestetti, I.; Russo, S.; Cereda, A.; Selicorni, A.; Larizza, L.; Finelli, P. Familial intragenic duplication of ANKRD11 underlying three patients of KBG syndrome. Mol. Cytogenet. 2015, 8, 20. [Google Scholar] [CrossRef] [Green Version]
- Isrie, M.; Hendriks, Y.; Gielissen, N.; Sistermans, E.A.; Willemsen, M.H.; Peeters, H.; Vermeesch, J.R.; Kleefstra, T.; Van Esch, H. Haploinsufficiency of ANKRD11 causes mild cognitive impairment, short stature and minor dysmorphisms. Eur. J. Hum. Genet. 2011, 20, 131–133. [Google Scholar] [CrossRef]
- Sacharow, S.; Li, D.; Fan, Y.-S.; Tekin, M. Familial 16q24.3 microdeletion involving ANKRD11 causes a KBG-like syndrome. Am. J. Med Genet. Part A 2012, 158, 547–552. [Google Scholar] [CrossRef]
- Kucharczyk, M.; Kochański, A.; Jezela-Stanek, A.; Kugaudo, M.; Sielska-Rotblum, D.; Gutkowska, A.; Krajewska-Walasek, M. The First Case of a Patient with De novo Partial Distal 16q Tetrasomy and a Data’s Review. Am J Med Genet A. 2014, 164A, 2541–2550. [Google Scholar] [CrossRef]
- Falco, M.; Amabile, S.; Acquaviva, F. RAI1 gene mutations: Mechanisms of Smith-Magenis syndrome. Appl. Clin. Genet. 2017, 10, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Harony-Nicolas, H.; De Rubeis, S.; Kolevzon, A.; Buxbaum, J.D. Phelan McDermid Syndrome. J. Child Neurol. 2015, 30, 1861–1870. [Google Scholar] [CrossRef] [Green Version]
- Donalies, M.; Cramer, M.; Ringwald, M.; Starzinski-Powitz, A. Expression of M-cadherin, a member of the cadherin multigene family, correlates with differentiation of skeletal muscle cells. Proc. Natl. Acad. Sci. USA 1991, 88, 8024–8028. [Google Scholar] [CrossRef] [Green Version]
- Bhalla, K.; Luo, Y.; Buchan, T.; Beachem, M.A.; Guzauskas, G.F.; Ladd, S.; Bratcher, S.J.; Schroer, R.J.; Balsamo, J.; Dupont, B.R.; et al. Alterations in CDH15 and KIRREL3 in Patients with Mild to Severe Intellectual Disability. Am. J. Hum. Genet. 2008, 83, 703–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petit, F.; Plessis, G.; DeCamp, M.; Cuisset, J.-M.; Blyth, M.; Pendlebury, M.; Andrieux, J. 21q21 deletion involving NCAM2: Report of 3 cases with neurodevelopmental disorders. Eur. J. Med Genet. 2015, 58, 44–46. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, O.; Fischetto, R.; Palumbo, P.; Nicastro, F.; Papadia, F.; Zelante, L.; Carella, M. De novo microduplication of CHL1 in a patient with non-syndromic developmental phenotypes. Molecular Cytogenetics 2015, 8, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaheen, R.; Patel, N.; Shamseldin, H.; Alzahrani, F.; Al-Yamany, R.; ALMoisheer, A.; Ewida, N.; Anazi, S.; Alnemer, M.; Elsheikh, M.; et al. Accelerating Matchmaking of Novel Dysmorphology Syndromes Through Clinical and Genomic Characterization of a Large Cohort. Genet Med. 2016, 18, 686–695. [Google Scholar] [CrossRef] [Green Version]
- Shaheen, R.; Al-Salam, Z.; El-Hattab, A.W.; Alkuraya, F.S. The syndrome dysmorphic facies, renal agenesis, ambiguous genitalia, microcephaly, polydactyly and lissencephaly (DREAM-PL): Report of two additional patients. Am. J. Med Genet. Part A 2016, 170, 3222–3226. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Present Case | Decipher 275865 | Decipher 300593 | Decipher 271478 | Decipher 333548 | Decipher 392985 | Decipher 322843 | |
|---|---|---|---|---|---|---|---|
| Age at Last Clinical Assessment | 9 years | <1 year | 4 years | 8 years | 7 years | <1 year | 8 years |
| Gender | M | F | M | M | F | F | M |
| Chromosome | 16 | 16 | 16 | 16 | 16 | 16 | 16 |
| Cytoband | q24.2q24.3 | q24.2q24.3 | q24.3 | q24.2q24.3 | q24.2q24.3 | q24.1q24.3 | q24.1q24.3 |
| Type | Gain | Gain | Gain | Gain | Gain | Gain | Gain |
| Start (hg19, bp) | 87502161 | 88317010 | 88755341 | 88473369 | 87577020 | 85342500 | 84341219 |
| Stop (hg19, bp) | 89688617 | 89479707 | 89897010 | 90111263 | 90148393 | 90294753 | 90111263 |
| Size (Mb) | 2.19 | 1.16 | 1.14 | 1.64 | 2.57 | 4.95 | 5.77 |
| # genes | 38 | 25 | 32 | 51 | 57 | 81 | 90 |
| Inheritance | dn | dn | ND | ND | ND | dn | dn |
| Clinical Significance | LP | LP | LP | VUS | VUS | LP | P |
| Global Developmental Delay/Intellectual Disability | + | + | NR | NR | + | + | + |
| Delayed Speech/Language development | + | ND | NR | NR | - | ND | - |
| Behavioral Problems | - | ND | NR | NR | - | ND | + |
| Seizures | - | + | NR | NR | - | - | + |
| Dysmorphic Features | Narrow and sloping forehead, bulbous nose with slightly anteverted nares | Abnormal facial shape | NR | NR | - | High anterior hairline, narrow and sloping forehead, bulbous nose, prominent nasal bridge, aplasia/Hypoplasia of the earlobes, hypertelorism, Micrognathia | - |
| Hands and Feet Abnormalities | Pronounced fingerpads, bilateral clinodactyly of the fifth finger | - | NR | NR | - | Deep palmar crease, finger clinodactyly | - |
| Others congenital abnormalities | CDH | - | NR | NR | - | MCA | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palumbo, O.; Palumbo, P.; Di Muro, E.; Cinque, L.; Petracca, A.; Carella, M.; Castori, M. A Private 16q24.2q24.3 Microduplication in a Boy with Intellectual Disability, Speech Delay and Mild Dysmorphic Features. Genes 2020, 11, 707. https://doi.org/10.3390/genes11060707
Palumbo O, Palumbo P, Di Muro E, Cinque L, Petracca A, Carella M, Castori M. A Private 16q24.2q24.3 Microduplication in a Boy with Intellectual Disability, Speech Delay and Mild Dysmorphic Features. Genes. 2020; 11(6):707. https://doi.org/10.3390/genes11060707
Chicago/Turabian StylePalumbo, Orazio, Pietro Palumbo, Ester Di Muro, Luigia Cinque, Antonio Petracca, Massimo Carella, and Marco Castori. 2020. "A Private 16q24.2q24.3 Microduplication in a Boy with Intellectual Disability, Speech Delay and Mild Dysmorphic Features" Genes 11, no. 6: 707. https://doi.org/10.3390/genes11060707
APA StylePalumbo, O., Palumbo, P., Di Muro, E., Cinque, L., Petracca, A., Carella, M., & Castori, M. (2020). A Private 16q24.2q24.3 Microduplication in a Boy with Intellectual Disability, Speech Delay and Mild Dysmorphic Features. Genes, 11(6), 707. https://doi.org/10.3390/genes11060707