Development of a Multilocus Sequence Typing Scheme for Giardia intestinalis

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Analysis of New Loci Using WGS Data
2.1.1. Genetic Diversity of Housekeeping Genes
2.1.2. Phylogenetic Analysis and Recombination Signals
2.1.3. Population Structure
2.2. Analysis of New Loci Using G. intestinalis in Stool Samples
2.2.1. Stool Samples
2.2.2. Amplification of New Loci in DNA Samples
2.2.3. Genetic Diversity and Phylogenetic Reconstruction
3. Discussion
4. Materials and Methods
4.1. Selection of New Genetic Markers and Design of Primers
4.2. In Silico Evaluation of Ten Genetic Markers
4.3. Utility of Selected Loci for Typing
4.4. Phylogenetic Inferences and Recombination Signals
4.5. Indices of Diversity and Genetic Structure
4.6. Assays from Human Stool Samples
4.6.1. Ethical Statement
4.6.2. Study Population, Detection and Typing of G. intestinalis
4.6.3. DNA Marker Assay of Stool Samples
4.6.4. Phylogenetic Reconstructions and Diversity Indices from Stool Samples
5. Conclusions
Availability of Data and Materials
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Haque, R. Human Intestinal Parasites. J. Health Popul. Nutr. 2007, 25, 387–391. [Google Scholar]
- CDC, Centers for Disease Control and Prevention. Parasites—Giardia. Available online: https://www.cdc.gov/parasites/giardia/infection-sources.html (accessed on 11 May 2020).
- Thompson, R.C.; Monis, P.T. Variation in Giardia: Implications for taxonomy and epidemiology. Adv. Parasitol. 2004, 58, 69–137. [Google Scholar] [CrossRef]
- Appelbee, A.J.; Thompson, R.C.; Olson, M.E. Giardia and Cryptosporidium in mammalian wildlife—Current status and future needs. Trends Parasitol. 2005, 21, 370–376. [Google Scholar] [CrossRef]
- Thompson, R.C.; Ash, A. Molecular epidemiology of Giardia and Cryptosporidium infections. Infect. Genet. Evol. 2016, 40, 315–323. [Google Scholar] [CrossRef] [Green Version]
- Abaza, S.M.; Sullivan, J.J.; Visvesvara, G.S. Isoenzyme profiles of four strains of Giardia lamblia and their infectivity to jirds. Am. J. Trop. Med. Hyg. 1991, 44, 63–68. [Google Scholar] [CrossRef]
- Andrews, R.H.; Adams, M.; Boreham, P.F.; Mayrhofer, G.; Meloni, B.P. Giardia intestinalis: Electrophoretic evidence for a species complex. Int. J. Parasitol. 1989, 19, 183–190. [Google Scholar] [CrossRef]
- Homan, W.L.; van Enckevort, F.H.; Limper, L.; van Eys, G.J.; Schoone, G.J.; Kasprzak, W.; Majewska, A.C.; van Knapen, F. Comparison of Giardia isolates from different laboratories by isoenzyme analysis and recombinant DNA probes. Parasitol. Res. 1992, 78, 316–323. [Google Scholar] [CrossRef]
- Nash, T. Surface antigen variability and variation in Giardia lamblia. Parasitol. Today 1992, 8, 229–234. [Google Scholar] [CrossRef]
- Weiss, J.B.; van Keulen, H.; Nash, T.E. Classification of subgroups of Giardia lamblia based upon ribosomal RNA gene sequence using the polymerase chain reaction. Mol. Biochem. Parasitol. 1992, 54, 73–86. [Google Scholar] [CrossRef]
- Wielinga, C.M.; Thompson, R.C. Comparative evaluation of Giardia duodenalis sequence data. Parasitology 2007, 134, 1795–1821. [Google Scholar] [CrossRef] [Green Version]
- Adam, R.D. Biology of Giardia lamblia. Clin. Microbiol. Rev. 2001, 14, 447–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Xiao, L. Zoonotic potential and molecular epidemiology of Giardia species and giardiasis. Clin. Microbiol. Rev. 2011, 24, 110–140. [Google Scholar] [CrossRef] [Green Version]
- Faria, C.P.; Zanini, G.M.; Dias, G.S.; da Silva, S.; Sousa, M.D.C. New multilocus genotypes of Giardia lamblia human isolates. Infect. Genet. Evol. 2017, 54, 128–137. [Google Scholar] [CrossRef]
- Read, C.M.; Monis, P.T.; Thompson, R.A. Discrimination of all genotypes of Giardia duodenalis at the glutamate dehydrogenase locus using PCR-RFLP. Infect. Genet. Evol. 2004, 4, 125–130. [Google Scholar] [CrossRef]
- Armson, A.; Yang, R.; Thompson, J.; Johnson, J.; Reid, S.; Ryan, U.M. Giardia genotypes in pigs in Western Australia: Prevalence and association with diarrhea. Exp. Parasitol. 2009, 121, 381–383. [Google Scholar] [CrossRef] [Green Version]
- Lebbad, M.; Mattsson, J.G.; Christensson, B.; Ljungström, B.; Backhans, A.; Andersson, J.O.; Svärd, S.G. From mouse to moose: Multilocus genotyping of Giardia isolates from various animal species. Vet. Parasitol. 2010, 168, 231–239. [Google Scholar] [CrossRef]
- Palmer, C.S.; Traub, R.J.; Robertson, I.D.; Devlin, G.; Rees, R.; Thompson, R.A. Determining the zoonotic significance of Giardia and Cryptosporidium in Australian dogs and cats. Vet. Parasitol. 2008, 154, 142–147. [Google Scholar] [CrossRef]
- Sprong, H.; Cacciò, S.M.; van der Giessen, J.W.B.; ZOOPNET Network and Partners. Identification of Zoonotic Genotypes of Giardia duodenalis. PLoS Negl. Trop. Dis. 2009, 3, e558. [Google Scholar] [CrossRef] [Green Version]
- Ryan, U.; Cacciò, S.M. Zoonotic potential of Giardia. Int. J. Parasitol. 2013, 43, 943–956. [Google Scholar] [CrossRef]
- Lee, M.F.; Auer, H.; Lindo, J.F.; Walochnik, J. Multilocus sequence analysis of Giardia spp. isolated from patients with diarrhea in Austria. Parasitol. Res. 2017, 116, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Plutzer, J.; Ongerth, J.; Karanis, P. Giardia taxonomy, phylogeny and epidemiology: Facts and open questions. Int. J. Hyg. Environ. Health 2010, 213, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, G.; Chen, G.; Jian, F.; Zhang, S.; Feng, C.; Wang, R.; Jinfeng, Z.; Dong, H.; Hua, J.; et al. Multilocus genotyping of Giardia duodenalis in dairy cattle in Henan, China. PLoS ONE 2014, 9, e100453. [Google Scholar] [CrossRef] [Green Version]
- Ankarklev, J.; Lebbad, M.; Einarsson, E.; Franzén, O.; Ahola, H.; Troell, K.; Svärd, S.G. A novel high-resolution multilocus sequence typing of Giardia intestinalis Assemblage a isolates reveals zoonotic transmission, clonal outbreaks and recombination. Infect. Genet. Evol. 2018, 60, 7–16. [Google Scholar] [CrossRef]
- Monis, P.T.; Andrews, R.H.; Mayrhofer, G.; Ey, P.L. Molecular systematics of the parasitic protozoan Giardia intestinalis. Mol. Biol. Evol. 1999, 16, 1135–1144. [Google Scholar] [CrossRef] [Green Version]
- Lasek-Nesselquist, E.; Mark Welch, D.M.; Sogin, M.L. The identification of a new Giardia duodenalis assemblage in marine vertebrates and a preliminary analysis of G. duodenalis population biology in marine systems. Int. J. Parasitol. 2010, 40, 1063–1074. [Google Scholar] [CrossRef] [Green Version]
- Poxleitner, M.K.; Carpenter, M.L.; Mancuso, J.J.; Wang, C.J.; Dawson, S.C.; Cande, W.Z. Evidence for karyogamy and exchange of genetic material in the binucleate intestinal parasite Giardia intestinalis. Science 2008, 319, 1530–1533. [Google Scholar] [CrossRef] [Green Version]
- Teodorovic, S.; Braverman, J.M.; Elmendorf, H.G. Unusually Low Levels of Genetic Variation among Giardia lamblia Isolates. Eukaryot. Cell 2007, 6, 1421–1430. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Jerlström-Hultqvist, J.; Andersson, J.O. Genome-wide analyses of recombination suggest that Giardia intestinalis assemblages represent different species. Mol. Biol. Evol. 2012, 29, 2895–2898. [Google Scholar] [CrossRef] [Green Version]
- Choy, S.H.; Mahdy, M.A.K.; Al-Mekhlafi, H.M.; Low, V.L.; Surin, J. Population expansion and gene flow in Giardia duodenalis as revealed by triosephosphate isomerase gene. Parasites Vectors 2015, 8, 454. [Google Scholar] [CrossRef] [Green Version]
- Durigan, M.; Ciampi-Guillardi, M.; Rodrigues, R.C.A.; Greinert-Goulart, J.A.; Siqueira-Castro, I.C.V.; Leal, D.A.G.; Yamashiro, S.; Bonatti, T.R.; Zucchi, M.I.; Franco, R.M.; et al. Population genetic analysis of Giardia duodenalis: Genetic diversity and haplotype sharing between clinical and environmental sources. MicrobiologyOpen 2017, 6, e00424. [Google Scholar] [CrossRef]
- Cooper, M.A.; Adam, R.D.; Worobey, M.; Sterling, C.R. Population genetics provides evidence for recombination in Giardia. Curr. Biol. 2007, 17, 1984–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urwin, R.; Maiden, M.C. Multi-locus sequence typing: A tool for global epidemiology. Trends Microbiol. 2003, 11, 479–487. [Google Scholar] [CrossRef]
- Sullivan, C.B.; Diggle, M.A.; Clarke, S.C. Multilocus sequence typing: Data analysis in clinical microbiology and public Health. Mol. Biotechnol. 2005, 29, 245–254. [Google Scholar] [CrossRef]
- Diosque, P.; Tomasini, N.; Lauthier, J.J.; Messenger, L.A.; Monje Rumi, M.M.; Ragone, P.G.; Alberti-D’Amato, A.M.; Pérez Brandán, C.; Barnabé, C.; Tibayrenc, M.; et al. Optimised multilocus sequence typing (MLST) scheme for Trypanosoma cruzi. PLoS Negl. Trop. Dis. 2014, 8, e3117. [Google Scholar] [CrossRef]
- Herrera, G.; Hernández, C.; Ayala, M.S.; Flórez, C.; Teherán, A.A.; Ramírez, J.D. Evaluation of a Multilocus Sequence Typing (MLST) scheme for Leishmania (Viannia) braziliensis and Leishmania (Viannia) panamensis in Colombia. Parasites Vectors 2017, 10, 236. [Google Scholar] [CrossRef]
- Ramírez, J.D.; Tapia-Calle, G.; Guhl, F. Genetic structure of Trypanosoma cruzi in Colombia revealed by a High-throughput Nuclear Multilocus Sequence Typing (nMLST) approach. BMC Genet. 2013, 14, 96. [Google Scholar] [CrossRef] [Green Version]
- Yeo, M.; Mauricio, I.L.; Messenger, L.A.; Lewis, M.D.; Llewellyn, M.S.; Acosta, N.; Bhattacharyya, T.; Diosque, P.; Carrasco, H.J.; Miles, M.A. Multilocus Sequence Typing (MLST) for Lineage Assignment and High Resolution Diversity Studies in Trypanosoma cruzi. PLoS Negl. Trop. Dis. 2011, 5, e1049. [Google Scholar] [CrossRef]
- Muñoz, M.; Camargo, M.; Ramírez, J.D. Estimating the Intra-taxa diversity, population genetic structure, and evolutionary pathways of Cryptococcus neoformans and Cryptococcus gattii. Front. Genet. 2018, 9, 148. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, M.; Wintaco, M.; Muñoz, S.; Ramírez, J.D. Dissecting the Heterogeneous Population Genetic Structure of Candida albicans: Limitations and Constraints of the Multilocus Sequence Typing Scheme. Front. Microbiol. 2019, 10, 1052. [Google Scholar] [CrossRef]
- Bougnoux, M.E.; Morand, S.; d’Enfert, C. Usefulness of multilocus sequence typing for characterisation of clinical isolates of Candida albicans. J. Clin. Microbiol. 2002, 40, 1290–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inouye, M.; Dashnow, H.; Raven, L.A.; Schultz, M.B.; Pope, B.J.; Tomita, T.; Zobel, J.; Holt, K.E. SRST2: Rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 2014, 6, 90. [Google Scholar] [CrossRef] [Green Version]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- Tomasini, N.; Lauthier, J.J.; Llewellyn, M.S.; Diosque, P. MLSTest: Novel software for multilocus sequence data analysis in eukaryotic organisms. Infect. Genet. Evol. 2013, 20, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Boil. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef] [Green Version]
- Rieux, A.; Halkett, F.; De Lapeyre de Bellaire, L.; Zapater, M.F.; Rousset, F.; Ravigné, V.; Carlier, J. Inferences on pathogenic fungus population structures from microsatellite data: New insights from spatial genetics approaches. Mol. Ecol. 2011, 20, 1661–1674. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Wright, S. Evolution and the Genetics of Populations: A Treatise in Four Volumes: Vol. 4: Variability within and among Natural Populations; University of Chicago Press: Chicago, IL, USA, 1978. [Google Scholar]
- Sánchez, A.; Muñoz, M.; Gómez, N.; Tabares, J.; Segura, L.; Salazar, Á.; Restrepo, C.; Ruíz, M.; Reyes, P.; Qian, Y.; et al. Molecular epidemiology of Giardia, Blastocystis and Cryptosporidium among indigenous children from the Colombian Amazon Basin. Front. Microbiol. 2017, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Higuera, A.; Villamizar, X.; Herrera, G.; Giraldo, J.C.; Vasquez-A, L.R.; Urbano, P.; Villalobos, O.; Tovar, C.; Ramírez, J.D. Molecular detection and genotyping of intestinal protozoa from different biogeographical regions of Colombia. PeerJ 2020, 8, e8554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, K.M.; Feil, E.J. The secret life of the multilocus sequence type. Int. J. Antimicrob. Agents 2007, 29, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Jerlström-Hultqvist, J.; Ankarklev, J.; Svärd, S.G. Is human giardiasis caused by two different Giardia species? Gut Microbes 2010, 1, 379–382. [Google Scholar] [CrossRef] [Green Version]
- Ankarklev, J.; Franzén, O.; Peirasmaki, D.; Jerlström-Hultqvist, J.; Lebbad, M.; Andersson, J.; Svärd, S.G. Comparative genomic analyses of freshly isolated Giardia intestinalis assemblage A isolates. BMC Genom. 2015, 16, 697. [Google Scholar] [CrossRef] [Green Version]
- Cacciò, S.M.; Beck, R.; Lalle, M.; Marinculic, A.; Pozio, E. Multilocus genotyping of Giardia duodenalis reveals striking differences between assemblages A and B. Int. J. Parasitol. 2008, 38, 1523–1531. [Google Scholar] [CrossRef]
- Blouin, M.S.; Yowell, C.A.; Courtney, C.H.; Dame, J.B. Host movement and the genetic structure of populations of parasitic nematodes. Genetics 1995, 141, 1007–1014. [Google Scholar]
- Miyoshi-Akiyama, T.; Hayakawa, K.; Ohmagari, N.; Shimojima, M.; Kirikae, T. Multilocus sequence typing (MLST) for characterisation of Enterobacter cloacae. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Chassain, B.; Lemée, L.; Didi, J.; Thiberge, J.M.; Brisse, S.; Pons, J.L.; Pestel-Caron, M. Multilocus sequence typing analysis of Staphylococcus lugdunensis implies a clonal population structure. J. Clin. Microbiol. 2012, 50, 3003–3009. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Losada, M.; Arenas, M.; Castro-Nallar, E. Multilocus sequence typing of pathogens. In Book Genetics and Evolution of Infectious Disease, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 383–404. [Google Scholar] [CrossRef]
- Minetti, C.; Lamden, K.; Durband, C.; Cheesbrough, J.; Fox, A.; Wastling, J.M. Determination of Giardia duodenalis assemblages and multilocus genotypes in patients with sporadic giardiasis from England. Parasites Vectors 2015, 8, 444. [Google Scholar] [CrossRef] [Green Version]
- Cacciò, S.M.; Ryan, U. Molecular epidemiology of giardiasis. Mol. Biochem. Parasitol. 2008, 160, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, M.L.; Assaf, Z.J.; Gourguechon, S.; Cande, W.Z. Nuclear inheritance and genetic exchange without meiosis in the binucleate parasite Giardia intestinalis. J. Cell Sci. 2012, 125, 2523–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birky, C.W., Jr. Sex: Is Giardia doing it in the dark? Curr. Biol. 2005, 15, R56–R58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, M.W.; Sunday, J. Things fall apart: Biological species form unconnected parsimony networks. Biol. Lett. 2007, 3, 509–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarcz, S.; Przyboś, E.; Surmacz, M. An assessment of haplotype variation in ribosomal and mitochondrial DNA fragments suggests incomplete lineage sorting in some species of the Paramecium aurelia complex (Ciliophora, Protozoa). Mol. Phylogenet. Evol. 2013, 67, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Detwiler, J.T.; Criscione, C.D. An infectious topic in reticulate evolution: Introgression and hybridisation in animal parasites. Genes 2010, 1, 102–123. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Collins, L.J. Reconstruction of sugar metabolic pathways of Giardia lamblia. Int. J. Proteom. 2012, 2012, 980829. [Google Scholar] [CrossRef] [Green Version]
- Hernández, Y.; Shpak, M.; Duarte, T.T.; Mendez, T.L.; Maldonado, R.A.; Roychowdhury, S.; Rodrigues, M.L.; Das, S. Novel role of sphingolipid synthesis genes in regulating giardial encystation. Infect. Immun. 2008, 76, 2939–2949. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patiño, L.H.; Camargo, M.; Muñoz, M.; Ríos-Chaparro, D.I.; Patarroyo, M.A.; Ramírez, J.D. Unveiling the multilocus sequence typing (MLST) schemes and core genome phylogenies for genotyping Chlamydia trachomatis. Front. Microbiol. 2018, 9, 1854. [Google Scholar] [CrossRef]
- Martin, D.P.; Williamson, C.; Posada, D. RDP2: Recombination detection and analysis from sequence alignments. Bioinformatics 2005, 21, 260–262. [Google Scholar] [CrossRef]






{kind=link}
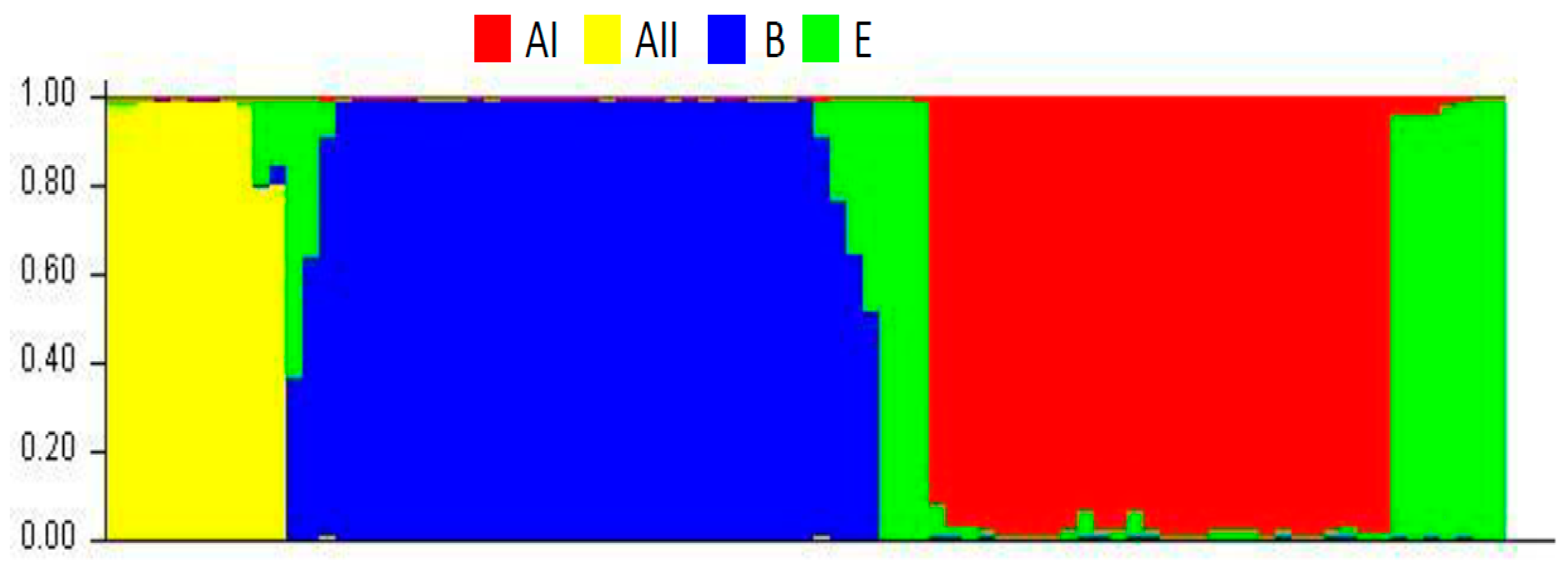{kind=link}
{kind=link}
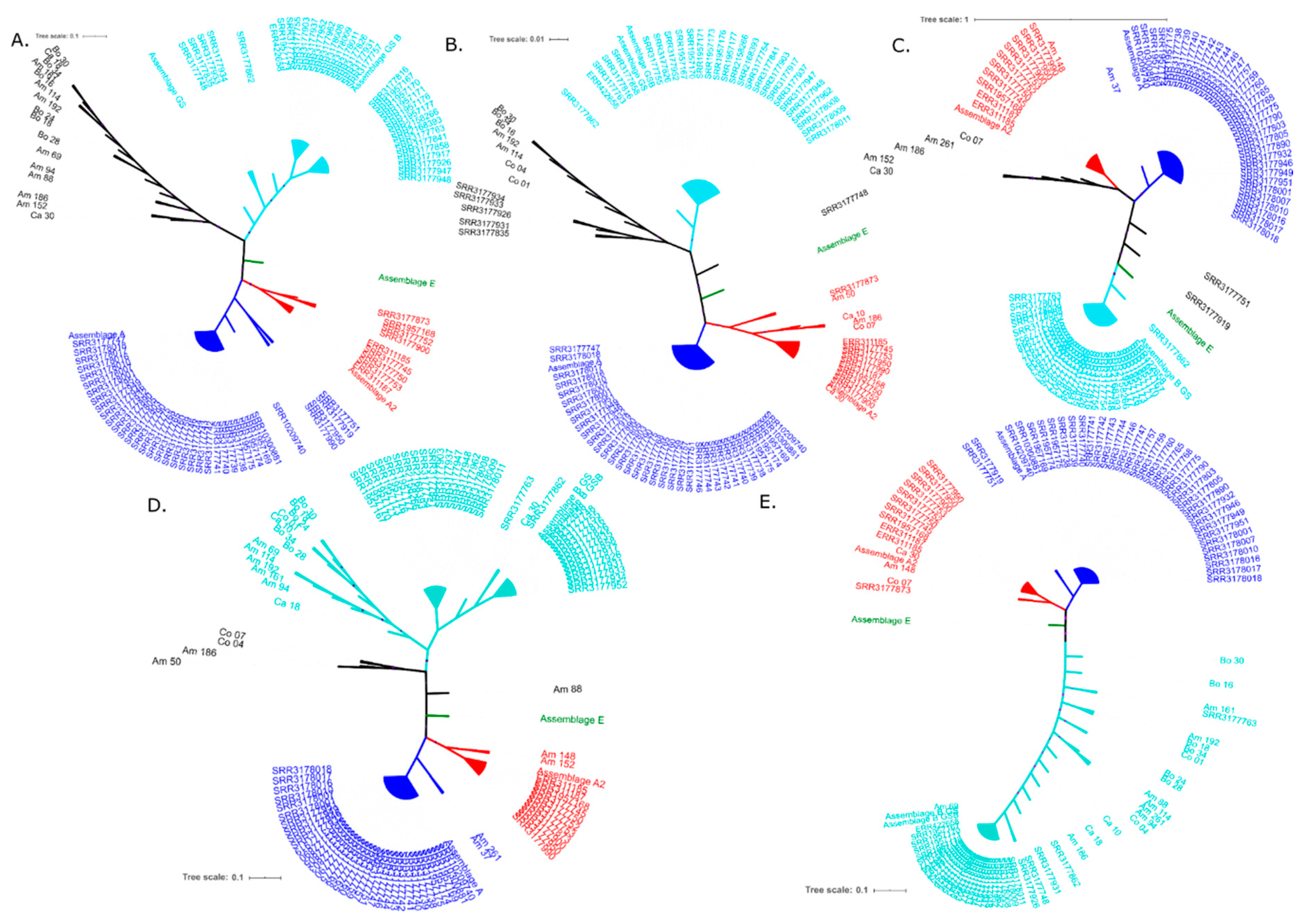{kind=link}
{kind=link}
| Marker | No. of Nucleotide Sites | No. of Sequences | Total Number of Sites | S | h | Hd (SD) | Pi (SD) | Theta | Tajima’s D test | Rm | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| WGS data consensus sequences | Concatenated | 11978 | 85 | 11496 | 2651 | 34 | 0.842 (0.036) | 0.086 (0.003) | 0.0488 | 2.676 * | 191 |
| ACS | 2190 | 85 | 1857 | 394 | 19 | 0.81 (0.032) | 0.078 (0.002) | 0.0451 | 2.550 * | 30 | |
| Enolase | 1338 | 85 | 1252 | 267 | 13 | 0.761 (0.036) | 0.071 (0.003) | 0.0455 | 1.983 | 16 | |
| FBA | 972 | 85 | 937 | 180 | 12 | 0.762 (0.03) | 0.07 (0.002) | 0.0423 | 2.242 * | 11 | |
| PFP-ALHA1 | 1650 | 85 | 1635 | 392 | 15 | 0.756 (0.034) | 0.092 (0.003) | 0.05 | 2.911 ** | 23 | |
| PGK | 1230 | 85 | 1204 | 272 | 16 | 0.799 (0.033) | 0.091 (0.002) | 0.047 | 3.169 ** | 22 | |
| GDH | 1386 | 85 | 1345 | 224 | 16 | 0.781 (0.036) | 0.058 (0.002) | 0.034 | 2.362 * | 17 | |
| NADP-ME | 1689 | 85 | 1625 | 464 | 15 | 0.791 (0.034) | 0.107 (0.004) | 0.06 | 2.641 * | 42 | |
| SPT | 1665 | 85 | 1641 | 458 | 11 | 0.686 (0.031) | 0.108 (0.003) | 0.059 | 2.896 ** | 24 | |
| TPI | 774 | 85 | 774 | 194 | 14 | 0.74 (0.034) | 0.095 (0.003) | 0.053 | 2.718 ** | 24 | |
| Sequences obtained from stool samples | ACS | 562 | 101 | 297 | 278 | 26 | 0.831 (0.025) | 0.21 (0.027) | 0.326 | –1.201 | 38 |
| Enolase | 428 | 97 | 350 | 162 | 19 | 0.756 (0.029) | 0.077 (0.005) | 0.108 | –0.978 | 26 | |
| GDH | 365 | 108 | 227 | 192 | 21 | 0.716 (0.039) | 0.091 (0.013) | 0.249 | –2.118 ** | 26 | |
| SPT | 487 | 91 | 175 | 134 | 10 | 0.579 (0.031) | 0.114 (0.013) | 0.221 | –1.639 | 15 | |
| TPI | 450 | 106 | 448 | 133 | 21 | 0.793 (0.026) | 0.109 (0.002) | 0.061 | 2.531 * | 52 |
| Name | ACS | Enolase | FBA | PFP-ALPHA1 | PGK | GDH | NADP-ME | SPT | TPI | All Loci |
|---|---|---|---|---|---|---|---|---|---|---|
| Number of Alleles | 21 | 15 | 14 | 15 | 18 | 20 | 15 | 11 | 14 | 53 |
| Number of Polymorphisms | 727 | 353 | 215 | 407 | 298 | 265 | 528 | 482 | 194 | 3469 |
| Typing Efficiency | 0.029 | 0.042 | 0.065 | 0.037 | 0.06 | 0.075 | 0.028 | 0.023 | 0.072 | 0.041 |
| DP (95% Confidence Interval) | 0.815 (0.748–0.881) | 0.768 (0.693–0.843) | 0.775 (0.715–0.835) | 0.756 (0.688–0.824) | 0.801 (0.732–0.869) | 0.796 (0.72–0.873) | 0.791 (0.722–0.861) | 0.686 (0.623–0.749) | 0.74 (0.671–0.81) | 0.885 (0.816–0.955) |
| Event No. | Found in | Recombinants | Major Parent | Minor Parent | Detection Methods | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| RDP | GENE CONV | BootScan | MaxiChi | Chimaera | SiScan | 3Seq | |||||
| 1 | 1 | SRR3177873 | SRR317790 | SRR3177948 | + | + | + | + | + | + | + |
| 2 | 1 | SRR3177873 | SRR3177900 | ERR422658 | + | + | + | + | + | + | + |
| 3 | 2 | Assemblage_A2 | SRR317799 | SRR3177750 | - | + | - | + | - | + | + |
| 4 | 1 | ERR422658 | SRR3177816 | Unknown | - | + | - | + | + | + | + |
| 5 | 1 | SRR3177873 | SRR3177900 | ERR422658 | - | + | - | - | - | - | - |
| 6 | 1 | SRR3177873 | SRR317790 | Unknown | - | + | + | - | - | - | - |
| 7 | 1 | SRR3177751 | SRR317791 | Unknown | - | - | - | + | - | - | - |
| 8 | 4 | SRR3177750 | SRR317790 | Unknown | + | + | + | + | + | + | + |
| 9 | 2 | SRR3177950 | SRR317790 | SRR3177919 | - | + | - | + | + | - | - |
| 10 | 1 | SRR3177873 | SRR3177950 | ERR422658 | - | + | - | - | - | - | - |
| 11 | 1 | SRR3177757 | SRR102097 | ERR422658 | - | + | + | + | + | + | + |
| 12 | 1 | SRR3177757 | SRR102097 | SRR3177816 | - | + | - | + | + | + | + |
| 13 | 10 | SRR3177948 | SRR3178011 | SRR3177926 | - | - | - | + | + | - | + |
| 14 | 1 | SRR3177862 | Assemblage_B | Unknown | - | - | - | + | + | - | + |
| 15 | 1 | SRR3177931 | Unknown | SRR3178011 | - | - | - | + | - | - | + |
| 16 | 5 | SRR3177952 | SRR3178011 | SRR3177926 | - | - | - | + | - | - | + |
| 17 | 1 | SRR3177763 | Unknown | SRR3177926 | + | - | - | + | + | + | + |
| Assemblages | Hs | Ks | Kxy | Gst | DeltaSt | GammaSt | Nst | Fst | Dxy | Da | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Concatenated | AI | AII | 0.353 | 1.616 | 13.844 | 0.344 | 0.004 | 0.760 | 0.809 | 0.808 | 0.012 | 0.009 |
| AI | B | 0.536 | 7.910 | 193.217 | 0.301 | 0.078 | 0.923 | 0.962 | 0.961 | 0.162 | 0.156 | |
| AII | B | 0.909 | 12.207 | 194.079 | 0.038 | 0.061 | 0.951 | 0.951 | 0.948 | 0.163 | 0.155 | |
| ACS | AI | AII | 0.300 | 3.952 | 36.542 | 0.421 | 0.006 | 0.774 | 0.822 | 0.822 | 0.018 | 0.015 |
| AI | B | 0.435 | 3.732 | 298.743 | 0.396 | 0.075 | 0.976 | 0.989 | 0.988 | 0.151 | 0.149 | |
| AII | B | 0.757 | 8.637 | 301.443 | 0.115 | 0.060 | 0.935 | 0.971 | 0.967 | 0.152 | 0.147 | |
| Enolase | AI | AII | 0.192 | 0.689 | 12.086 | 0.533 | 0.003 | 0.858 | 0.947 | 0.947 | 0.010 | 0.009 |
| AI | B | 0.306 | 1.244 | 177.760 | 0.516 | 0.069 | 0.986 | 0.993 | 0.993 | 0.141 | 0.140 | |
| AII | B | 0.561 | 1.532 | 178.279 | 0.241 | 0.055 | 0.979 | 0.994 | 0.993 | 0.142 | 0.141 | |
| FBA | AI | AII | 0.083 | 0.426 | 10.610 | 0.799 | 0.004 | 0.905 | 0.932 | 0.932 | 0.011 | 0.011 |
| AI | B | 0.349 | 4.646 | 124.082 | 0.485 | 0.064 | 0.930 | 0.963 | 0.963 | 0.132 | 0.128 | |
| AII | B | 0.526 | 7.123 | 127.157 | 0.307 | 0.049 | 0.869 | 0.960 | 0.959 | 0.136 | 0.130 | |
| PFP-ALPHAI | AI | AII | 0.213 | 0.736 | 16.048 | 0.526 | 0.004 | 0.896 | 0.919 | 0.918 | 0.010 | 0.009 |
| AI | B | 0.176 | 1.496 | 295.903 | 0.695 | 0.089 | 0.990 | 0.995 | 0.994 | 0.181 | 0.180 | |
| AII | B | 0.424 | 3.025 | 293.796 | 0.333 | 0.077 | 0.977 | 0.991 | 0.990 | 0.180 | 0.178 | |
| PGK | AI | AII | 0.138 | 0.795 | 9.667 | 0.675 | 0.002 | 0.780 | 0.950 | 0.950 | 0.008 | 0.008 |
| AI | B | 0.467 | 2.942 | 216.423 | 0.366 | 0.088 | 0.974 | 0.988 | 0.987 | 0.178 | 0.176 | |
| AII | B | 0.651 | 3.995 | 217.676 | 0.233 | 0.052 | 0.942 | 0.990 | 0.989 | 0.179 | 0.177 | |
| GDH | AI | AII | 0.251 | 1.342 | 18.435 | 0.443 | 0.004 | 0.829 | 0.876 | 0.875 | 0.014 | 0.012 |
| AI | B | 0.402 | 1.580 | 152.200 | 0.423 | 0.056 | 0.980 | 0.990 | 0.989 | 0.113 | 0.112 | |
| AII | B | 0.724 | 3.149 | 150.617 | 0.127 | 0.042 | 0.949 | 0.979 | 0.977 | 0.112 | 0.109 | |
| NADP-ME | AI | AII | 0.181 | 2.074 | 15.462 | 0.572 | 0.003 | 0.703 | 0.745 | 0.745 | 0.010 | 0.007 |
| AI | B | 0.397 | 5.064 | 340.229 | 0.432 | 0.103 | 0.972 | 0.987 | 0.985 | 0.209 | 0.206 | |
| AII | B | 0.702 | 9.445 | 342.710 | 0.159 | 0.078 | 0.933 | 0.978 | 0.974 | 0.211 | 0.205 | |
| SPT | AI | AII | 0.167 | 1.702 | 22.629 | 0.603 | 0.005 | 0.827 | 0.858 | 0.858 | 0.014 | 0.012 |
| AI | B | 0.110 | 0.423 | 350.307 | 0.801 | 0.107 | 0.998 | 0.999 | 0.999 | 0.213 | 0.213 | |
| AII | B | 0.246 | 2.125 | 349.750 | 0.493 | 0.079 | 0.985 | 0.991 | 0.990 | 0.213 | 0.211 | |
| TPI | AI | AII | 0.137 | 0.178 | 6.500 | 0.641 | 0.003 | 0.935 | 0.947 | 0.946 | 0.008 | 0.008 |
| AI | B | 0.245 | 0.701 | 146.086 | 0.607 | 0.094 | 0.991 | 0.996 | 0.995 | 0.189 | 0.188 | |
| AII | B | 0.513 | 1.222 | 144.529 | 0.257 | 0.071 | 0.979 | 0.994 | 0.993 | 0.187 | 0.185 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Higuera, A.; Muñoz, M.; López, M.C.; Reyes, P.; Urbano, P.; Villalobos, O.; Ramírez, J.D. Development of a Multilocus Sequence Typing Scheme for Giardia intestinalis. Genes 2020, 11, 764. https://doi.org/10.3390/genes11070764
Higuera A, Muñoz M, López MC, Reyes P, Urbano P, Villalobos O, Ramírez JD. Development of a Multilocus Sequence Typing Scheme for Giardia intestinalis. Genes. 2020; 11(7):764. https://doi.org/10.3390/genes11070764
Chicago/Turabian StyleHiguera, Adriana, Marina Muñoz, Myriam Consuelo López, Patricia Reyes, Plutarco Urbano, Oswaldo Villalobos, and Juan David Ramírez. 2020. "Development of a Multilocus Sequence Typing Scheme for Giardia intestinalis" Genes 11, no. 7: 764. https://doi.org/10.3390/genes11070764
APA StyleHiguera, A., Muñoz, M., López, M. C., Reyes, P., Urbano, P., Villalobos, O., & Ramírez, J. D. (2020). Development of a Multilocus Sequence Typing Scheme for Giardia intestinalis. Genes, 11(7), 764. https://doi.org/10.3390/genes11070764