Convergence of Prognostic Gene Signatures Suggests Underlying Mechanisms of Human Prostate Cancer Progression

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. The You et al. Discovery Cohort (DISC)
2.2. Validation Datasets
2.3. Replicating You et al. Analysis
2.3.1. Pathway Activation Z-Score
2.3.2. Non-Negative Matrix Factorization
2.3.3. NNMF Random Forest Classifier
2.4. Replicating the Ramos-Montoya Classifier
2.5. Replicating the Prolaris Classifier
2.6. LPD (Latent Process Decomposition) DESNT
2.7. Statistical Analysis
3. Results
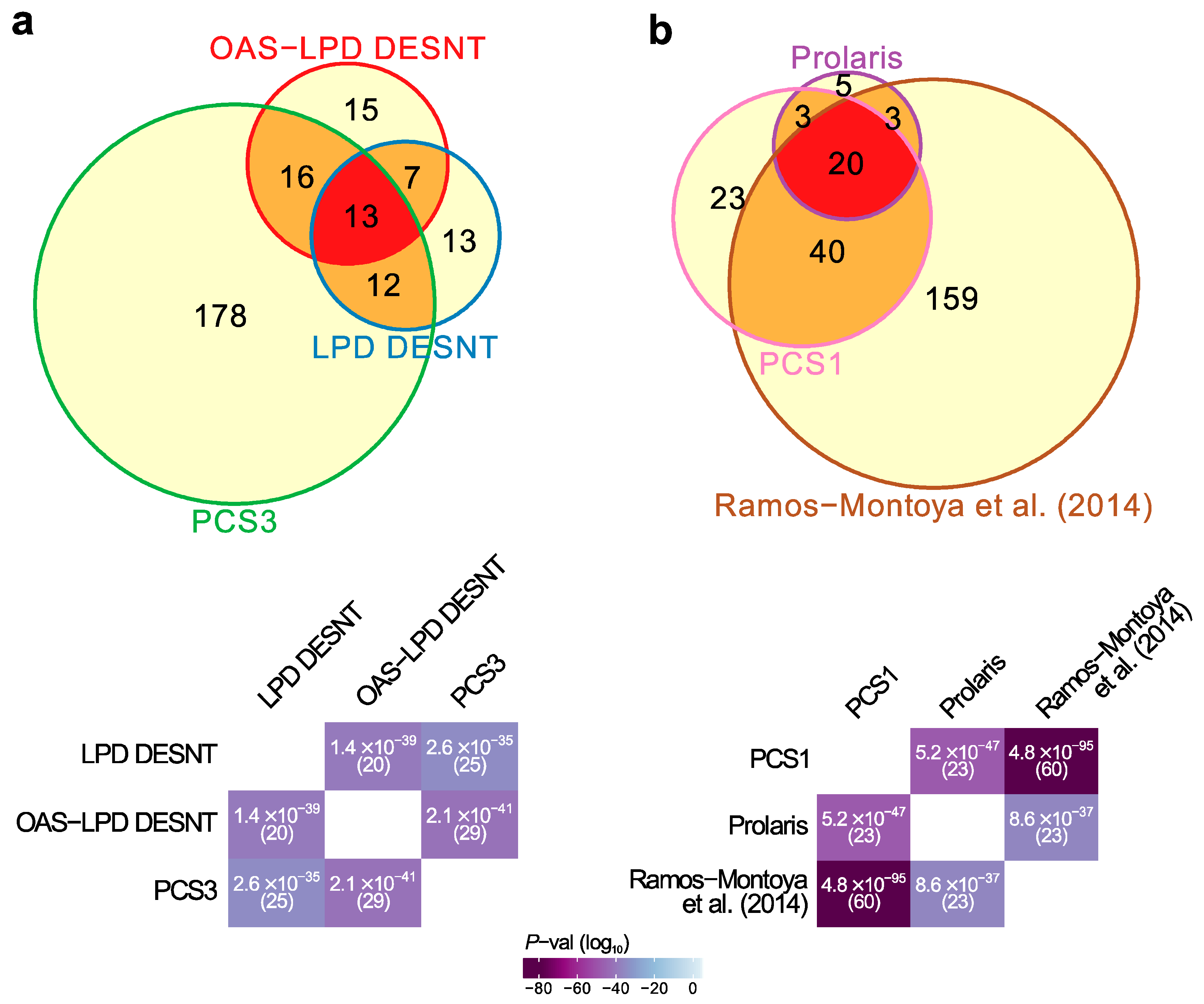
3.1. Relationships between Prostate Cancer Signatures
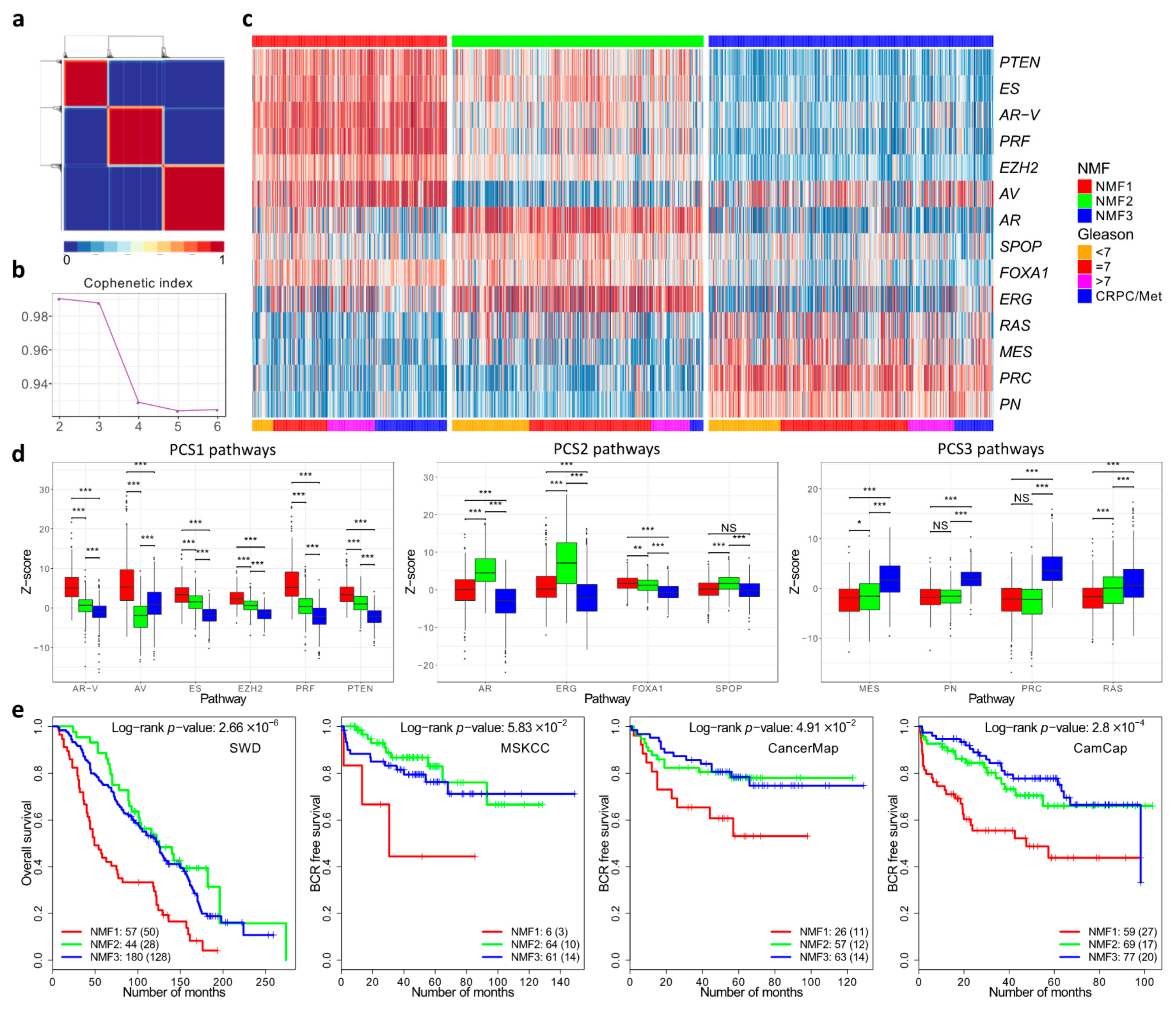
3.2. Cancer Subgroups Identified by Non-Negative Matrix Factorisation of Control Pathways
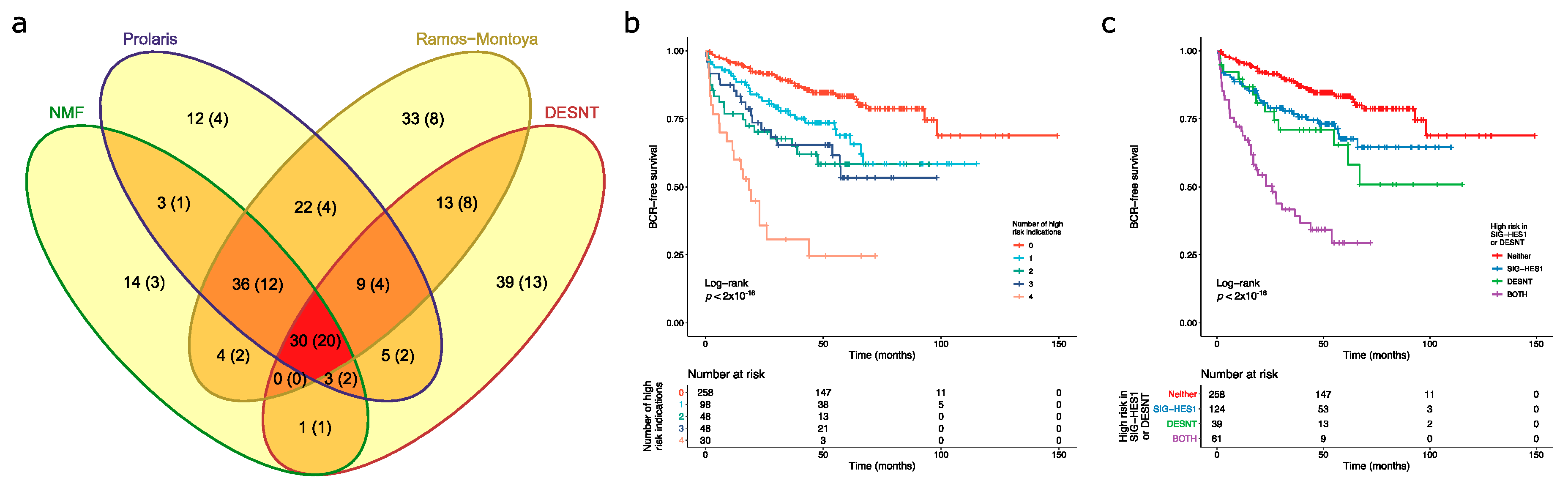
3.3. Overlaps in the Detection of Cancers at High Risk of PSA Failure
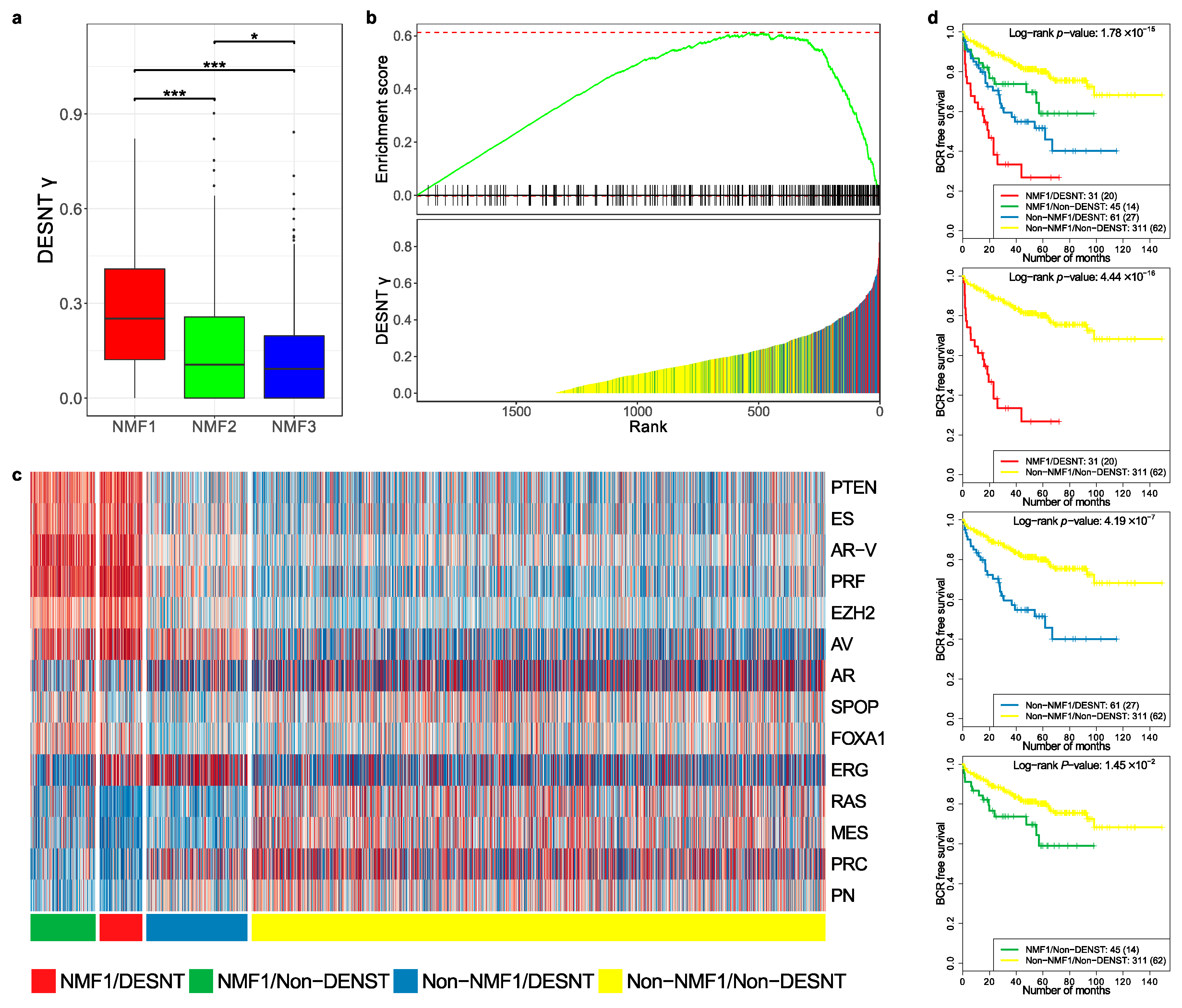
3.4. Comparison of DESNT and Non-Negative Matrix Factorisation Subgroups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- D’Amico, A.V.; Moul, J.; Carroll, P.R.; Sun, L.; Lubeck, D.; Chen, M.-H. Cancer-Specific Mortality After Surgery or Radiation for Patients With Clinically Localized Prostate Cancer Managed During the Prostate-Specific Antigen Era. J. Clin. Oncol. 2016, 21, 2163–2172. [Google Scholar]
- Buyyounouski, M.K.; Pickles, T.; Kestin, L.L.; Allison, R.; Williams, S.G. Validating the Interval to Biochemical Failure for the Identification of Potentially Lethal Prostate Cancer. J. Clin. Oncol. 2012, 30, 1857–1863. [Google Scholar] [CrossRef] [PubMed]
- Cucchiara, V.; Cooperberg, M.R.; Dall′Era, M.; Lin, D.W.; Montorsi, F.; Schalken, J.A.; Evans, C.P. Genomic Markers in Prostate Cancer Decision Making. Eur. Urol. 2018, 73, 572–582. [Google Scholar] [CrossRef]
- Agell, L.; Hernández, S.; Nonell, L.; Lorenzo, M.; Puigdecanet, E.; de Muga, S.; Juanpere, N.; Bermudo, R.; Fernández, P.L.; Lorente, J.A.; et al. 12-gene expression signature is associated with aggressive histological in prostate cancer: SEC14L1 and TCEB1 genes are potential markers of progression. Am. J. Pathol. 2012, 181, 1585–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibikova, M.; Chudin, E.; Arsanjani, A.; Zhou, L.; Garcia, E.W.; Modder, J.; Kostelec, M.; Barker, D.; Downs, T.; Fan, J.-B.; et al. Expression signatures that correlated with Gleason score and relapse in prostate cancer. Genomics 2007, 89, 666–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bismar, T.A.; Demichelis, F.; Riva, A.; Kim, R.; Varambally, S.; He, L.; Kutok, J.; Aster, J.C.; Tang, J.; Kuefer, R.; et al. Defining aggressive prostate cancer using a 12-gene model. Neoplasia 2006, 8, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Long, Q.; Johnson, B.A.; Osunkoya, A.O.; Lai, Y.-H.; Zhou, W.; Abramovitz, M.; Xia, M.; Bouzyk, M.B.; Nam, R.K.; Sugar, L.; et al. Protein-coding and microRNA biomarkers of recurrence of prostate cancer following radical prostatectomy. Am. J. Pathol. 2011, 179, 46–54. [Google Scholar] [CrossRef]
- Erho, N.; Crisan, A.; Vergara, I.A.; Mitra, A.P.; Ghadessi, M.; Buerki, C.; Bergstralh, E.J.; Kollmeyer, T.; Fink, S.; Haddad, Z.; et al. Discovery and Validation of a Prostate Cancer Genomic Classifier that Predicts Early Metastasis Following Radical Prostatectomy. PLoS ONE 2013, 8, e66855. [Google Scholar] [CrossRef] [Green Version]
- Glinsky, G.V.; Glinskii, A.B.; Stephenson, A.J.; Hoffman, R.M.; Gerald, W.L. Gene expression profiling predicts clinical outcome of prostate cancer. J. Clin. Investig. 2004, 113, 913–923. [Google Scholar] [CrossRef]
- Klein, E.A.; Cooperberg, M.R.; Magi-Galluzzi, C.; Simko, J.P.; Falzarano, S.M.; Maddala, T.; Chan, J.M.; Li, J.; Cowan, J.E.; Tsiatis, A.C.; et al. A 17-gene assay to predict prostate cancer aggressiveness in the context of Gleason grade heterogeneity, tumor multifocality, and biopsy undersampling. Eur. Urol. 2014, 66, 550–560. [Google Scholar] [CrossRef] [Green Version]
- Planche, A.; Bacac, M.; Provero, P.; Fusco, C.; Delorenzi, M.; Stehle, J.-C.; Stamenkovic, I. Identification of prognostic molecular features in the reactive stroma of human breast and prostate cancer. PLoS ONE 2011, 6, e18640. [Google Scholar] [CrossRef] [PubMed]
- Rajan, P.; Stockley, J.; Sudbery, I.M.; Fleming, J.T.; Hedley, A.; Kalna, G.; Sims, D.; Ponting, C.P.; Heger, A.; Robson, C.N.; et al. Identification of a candidate prognostic gene signature by transcriptome analysis of matched pre- and post-treatment prostatic biopsies from patients with advanced prostate cancer. BMC Cancer 2014, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, S.; Ross, K.N.; Lander, E.S.; Golub, T.R. A molecular signature of metastasis in primary solid tumors. Nat. Genet. 2003, 33, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Febbo, P.G.; Ross, K.; Jackson, D.G.; Manola, J.; Ladd, C.; Tamayo, P.; Renshaw, A.A.; D’Amico, A.V.; Richie, J.P.; et al. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell 2002, 1, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Varambally, S.; Yu, J.; Laxman, B.; Rhodes, D.R.; Mehra, R.; Tomlins, S.A.; Shah, R.B.; Chandran, U.; Monzon, F.A.; Becich, M.J.; et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 2005, 8, 393–406. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-L.; Schroeder, B.E.; Ma, X.-J.; Cutie, C.J.; Wu, S.; Salunga, R.; Zhang, Y.; Kattan, M.W.; Schnabel, C.A.; Erlander, M.G.; et al. Development and validation of a 32-gene prognostic index for prostate cancer progression. Proc. Natl. Acad. Sci. USA 2013, 110, 6121–6126. [Google Scholar] [CrossRef] [Green Version]
- Mo, F.; Lin, D.; Takhar, M.; Ramnarine, V.R.; Dong, X.; Bell, R.H.; Volik, S.V.; Wang, K.; Xue, H.; Wang, Y.; et al. Stromal Gene Expression is Predictive for Metastatic Primary Prostate Cancer. Eur. Urol. 2018, 73, 524–532. [Google Scholar] [CrossRef]
- Yu, J.; Yu, J.; Rhodes, D.R.; Tomlins, S.A.; Cao, X.; Chen, G.; Mehra, R.; Wang, X.; Ghosh, D.; Shah, R.B.; et al. A polycomb repression signature in metastatic prostate cancer predicts cancer outcome. Cancer Res. 2007, 67, 10657–10663. [Google Scholar] [CrossRef] [Green Version]
- Cuzick, J.; Swanson, G.P.; Fisher, G.; Brothman, A.R.; Berney, D.M.; Reid, J.E.; Mesher, D.; Speights, V.O.; Stankiewicz, E.; Foster, C.S.; et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: A retrospective study. Lancet Oncol. 2011, 12, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Irshad, S.; Bansal, M.; Castillo-Martin, M.; Zheng, T.; Aytes, A.; Wenske, S.; Le Magnen, C.; Guarnieri, P.; Sumazin, P.; Benson, M.C.; et al. A molecular signature predictive of indolent prostate cancer. Sci. Transl. Med. 2013, 5, 202ra122. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.L.; Massie, C.E.; Ramos-Montoya, A.; Zecchini, V.; Scott, H.E.; Lamb, A.D.; MacArthur, S.; Stark, R.; Warren, A.Y.; Mills, I.G.; et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell 2013, 23, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Montoya, A.R.; Lamb, A.D.; Russell, R.; Carroll, T.; Jurmeister, S.; Dalmau, N.G.; Massie, C.E.; Boren, J.; Bon, H.; Theodorou, V.; et al. HES6 drives a critical AR transcriptional programme to induce castration-resistant prostate cancer through activation of an E2F1-mediated cell cycle network. EMBO Mol. Med. 2014, 6, 651–661. [Google Scholar] [CrossRef] [Green Version]
- Luca, B.-A.; Brewer, D.S.; Edwards, D.R.; Edwards, S.; Whitaker, H.C.; Merson, S.; Dennis, N.; Cooper, R.A.; Hazell, S.; Warren, A.Y.; et al. DESNT: A Poor Prognosis Category of Human Prostate Cancer. Eur. Urol. Focus 2018, 4, 842–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross-Adams, H.; Lamb, A.D.; Dunning, M.J.; Halim, S.; Lindberg, J.; Massie, C.M.; Egevad, L.A.; Russell, R.; Ramos-Montoya, A.; Vowler, S.L.; et al. CamCaP Study Group, Integration of copy number and transcriptomics provides risk stratification in prostate cancer: A discovery and validation cohort study. EBioMedicine 2015, 2, 1133–1144. [Google Scholar] [CrossRef] [Green Version]
- Walker, S.M.; Knight, L.A.; McCavigan, A.M.; Logan, G.E.; Berge, V.; Sherif, A.; Pandha, H.; Warren, A.Y.; Davidson, C.; Uprichard, A.; et al. Molecular Subgroup of Primary Prostate Cancer Presenting with Metastatic Biology. Eur. Urol. 2017, 72, 509–518. [Google Scholar] [CrossRef] [Green Version]
- You, S.; Knudsen, B.S.; Erho, N.; Alshalalfa, M.; Takhar, M.; Al-deen Ashab, H.; Davicioni, E.; Karnes, R.J.; Klein, E.A.; Den, R.B.; et al. Integrated Classification of Prostate Cancer Reveals a Novel Luminal Subtype with Poor Outcome. Cancer Res. 2016, 76, 4948–4958. [Google Scholar] [CrossRef] [Green Version]
- Luca, B.-A.; Moulton, V.; Ellis, C.; Edwards, D.R.; Campbell, C.; Cooper, R.A.; Clark, J.; Brewer, D.S.; Cooper, C.S. A novel stratification framework for predicting outcome in patients with prostate cancer. Br. J. Cancer 2020, 122, 1467–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalonde, E.; Ishkanian, A.S.; Sykes, J.; Fraser, M.; Ross-Adams, H.; Erho, N.; Dunning, M.J.; Halim, S.; Lamb, A.D.; Moon, N.C.; et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: A retrospective cohort study. Lancet Oncol. 2014, 15, 1521–1532. [Google Scholar] [CrossRef]
- Ein-Dor, L.; Kela, I.; Getz, G.; Givol, D.; Domany, E. Outcome signature genes in breast cancer: Is there a unique set? Bioinformatics 2005, 21, 171–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michiels, S.; Koscielny, S.; Hill, C. Prediction of cancer outcome with microarrays: A multiple random validation strategy. Lancet 2005, 365, 488–492. [Google Scholar] [CrossRef]
- Ein-Dor, L.; Zuk, O.; Domany, E. Thousands of samples are needed to generate a robust gene list for predicting outcome in cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 5923–5928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van’t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.M.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.H.; Dudoit, S.; Luu, P.; Lin, D.M.; Peng, V.; Ngai, J.; Speed, T.P. Normalization for cDNA microarray data: A robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002, 30, e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. Affy-analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Sboner, A.; Demichelis, F.; Calza, S.; Pawitan, Y.; Setlur, S.R.; Hoshida, Y.; Perner, S.; Adami, H.-O.; Fall, K.; Mucci, L.A.; et al. Molecular sampling of prostate cancer: A dilemma for predicting disease progression. BMC Med. Genom. 2010, 3, 8–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, D.M.; Haynor, D.R.; Castle, J.C.; Stepaniants, S.B.; Pellegrini, M.; Mao, M.; Johnson, J.M. Pathway and gene-set activation measurement from mRNA expression data: The tissue distribution of human pathways. Genome Biol. 2006, 7, R93. [Google Scholar] [CrossRef] [Green Version]
- Gaujoux, R.; Seoighe, C. A flexible R package for nonnegative matrix factorization. BMC Bioinform. 2010, 11, 367–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, S.; Girolami, M.; Campbell, C.; Breitling, R. The latent process decomposition of cDNA microarray data sets. IEEE/ACM Trans. Comput. Biol. Bioinform. 2005, 2, 143–156. [Google Scholar] [CrossRef]
- Carrivick, L.; Rogers, S.; Clark, J.; Campbell, C.; Girolami, M.; Cooper, C. Identification of prognostic signatures in breast cancer microarray data using Bayesian techniques. J. R. Soc. Interface 2006, 3, 367–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergushichev, A.A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. BioRxiv 2016, 060012. [Google Scholar]
- Edwards, S.; Campbell, C.; Flohr, P.; Shipley, J.; Giddings, I.; Te-Poele, R.; Dodson, A.; Foster, C.; Clark, J.; Jhavar, S.; et al. Expression analysis onto microarrays of randomly selected cDNA clones highlights HOXB13 as a marker of human prostate cancer. Br. J. Cancer 2005, 92, 376–381. [Google Scholar] [CrossRef] [Green Version]
- Ewing, C.M.; Ray, A.M.; Lange, E.M.; Zuhlke, K.A.; Robbins, C.M.; Tembe, W.D.; Wiley, K.E.; Isaacs, S.D.; Johng, D.; Wang, Y.; et al. Germline mutations in HOXB13 and prostate-cancer risk. N. Engl. J. Med. 2012, 366, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Rubin, M.A.; Zhou, M.; Dhanasekaran, S.M.; Varambally, S.; Barrette, T.R.; Sanda, M.G.; Pienta, K.J.; Ghosh, D.; Chinnaiyan, A.M. α-Methylacyl coenzyme A racemase as a tissue biomarker for prostate cancer. JAMA 2002, 287, 1662–1670. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Citation | Year | Genes | Type | Discovery Method | Name |
|---|---|---|---|---|---|
| Agell et al. | 2012 | 12 | A | Association to Gleason | - |
| Bibkova et al. | 2007 | 16 | A | Association to Gleason | - |
| Bismar et al. | 2006 | 12 | A | Benign vs. Cancer vs. Metastases | - |
| Cuzick et al. | 2011 | 31 | B | Cell Cycle Genes | Prolaris |
| Erho et al. | 2013 | 22 | A | Cancers with Different Progressions | DECIPHER |
| Glinksy et al. | 2004 | 11 | A | PSA Failure vs. No-failure | - |
| Irshad et al. | 2013 | 19 | B | Aging Genes Altered in Indolent Cancer | - |
| Klein et al. | 2014 | 17 | A | Association with Outcome | OncotypeDX |
| Lalonde et al. | 2014 | 276 | U | Genes within Copy Number Changes | - |
| Long et al. | 2011 | 13 | A | PSA failure vs. No failure | - |
| Luca et al. | 2017 | 45 | U | LPD | DESNT |
| Luca et al. | 2020 | 49 | U | OAS-LPD | OAS-DESNT |
| Mo et al. | 2018 | 93 | B + A | Stroma association to metastasis | - |
| Planche et al. | 2011 | 48 | A | Normal vs. Tumour differential gene expression in stroma | - |
| Rajan et al. | 2014 | 7 | A | Before and After ADT | - |
| Ramos-Montoya et al. | 2014 | 222 | B | Genes Controlled by HES6 | - |
| Ramaswamy et al. | 2003 | 17 | A | Metastases vs. Primary | - |
| Ross-Adams et al. | 2014 | 100 | U | Clustering of Variable Genes | - |
| Sharma et al. | 2013 | 16 | B | Androgen Receptor Regulated | - |
| Singh et al. | 2002 | 29 | A | Associated with Gleason | - |
| Varambally et al. | 2005 | 44 | A | Metastases vs. Primary | - |
| Walker et al. | 2017 | 70 | U + A | HCA and PLS Regression * | - |
| Wu et al. | 2013 | 32 | A | Associated with Outcome | - |
| You et al. | 2016 | 428 | U | NNMF of Control Pathways | PCS1, PCS2, PCS3 |
| Yu et al. | 2007 | 7 | B | Polycomb Repression Signature | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luca, B.-A.; Moulton, V.; Ellis, C.; Connell, S.P.; Brewer, D.S.; Cooper, C.S. Convergence of Prognostic Gene Signatures Suggests Underlying Mechanisms of Human Prostate Cancer Progression. Genes 2020, 11, 802. https://doi.org/10.3390/genes11070802
Luca B-A, Moulton V, Ellis C, Connell SP, Brewer DS, Cooper CS. Convergence of Prognostic Gene Signatures Suggests Underlying Mechanisms of Human Prostate Cancer Progression. Genes. 2020; 11(7):802. https://doi.org/10.3390/genes11070802
Chicago/Turabian StyleLuca, Bogdan-Alexandru, Vincent Moulton, Christopher Ellis, Shea P. Connell, Daniel S. Brewer, and Colin S. Cooper. 2020. "Convergence of Prognostic Gene Signatures Suggests Underlying Mechanisms of Human Prostate Cancer Progression" Genes 11, no. 7: 802. https://doi.org/10.3390/genes11070802
APA StyleLuca, B. -A., Moulton, V., Ellis, C., Connell, S. P., Brewer, D. S., & Cooper, C. S. (2020). Convergence of Prognostic Gene Signatures Suggests Underlying Mechanisms of Human Prostate Cancer Progression. Genes, 11(7), 802. https://doi.org/10.3390/genes11070802