Mutation Spectrum and De Novo Mutation Analysis in Stickler Syndrome Patients with High Myopia or Retinal Detachment


Abstract
:1. Introduction
2. Materials and Methods
3. Results
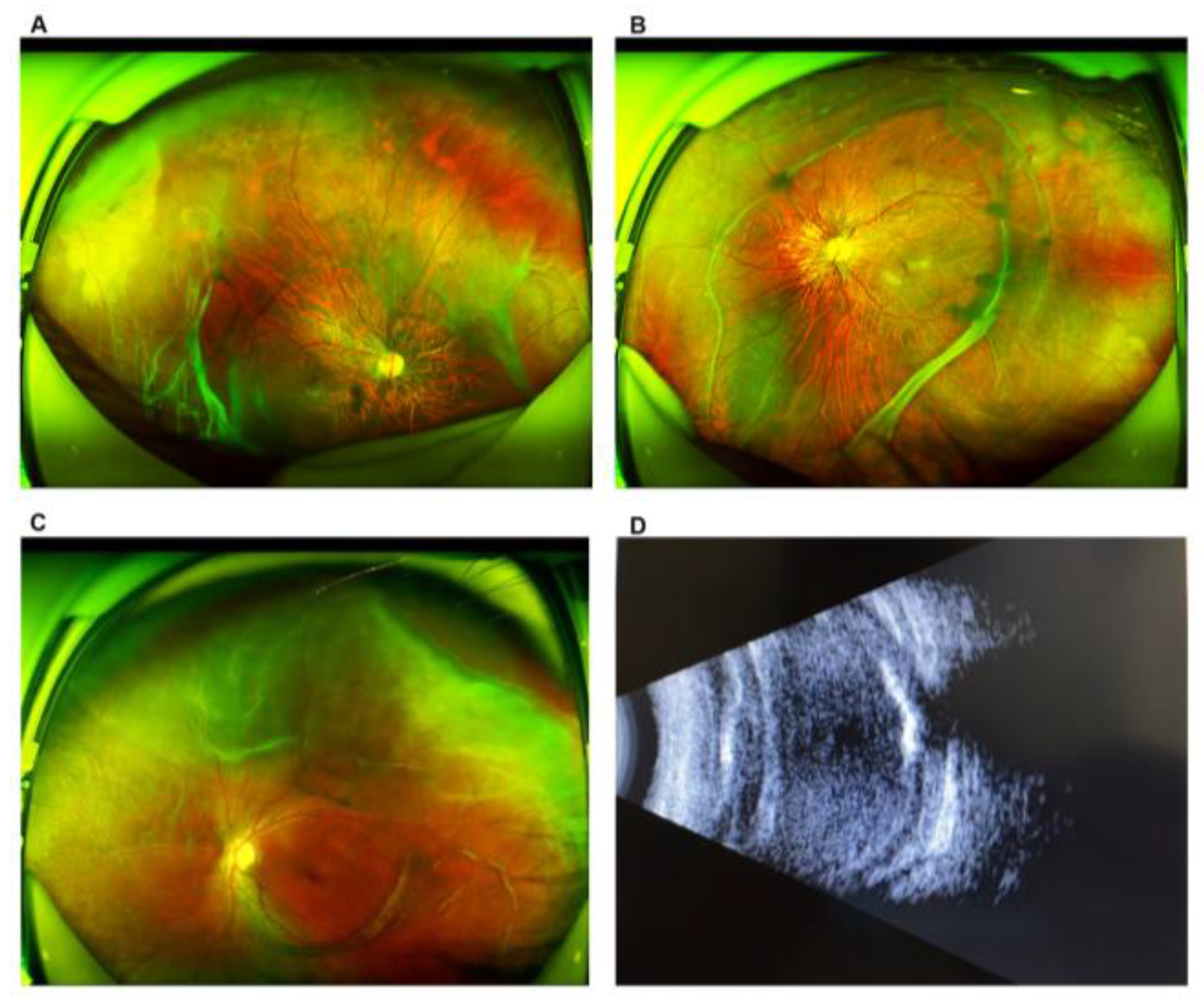
3.1. Demographic and Clinical Characteristics
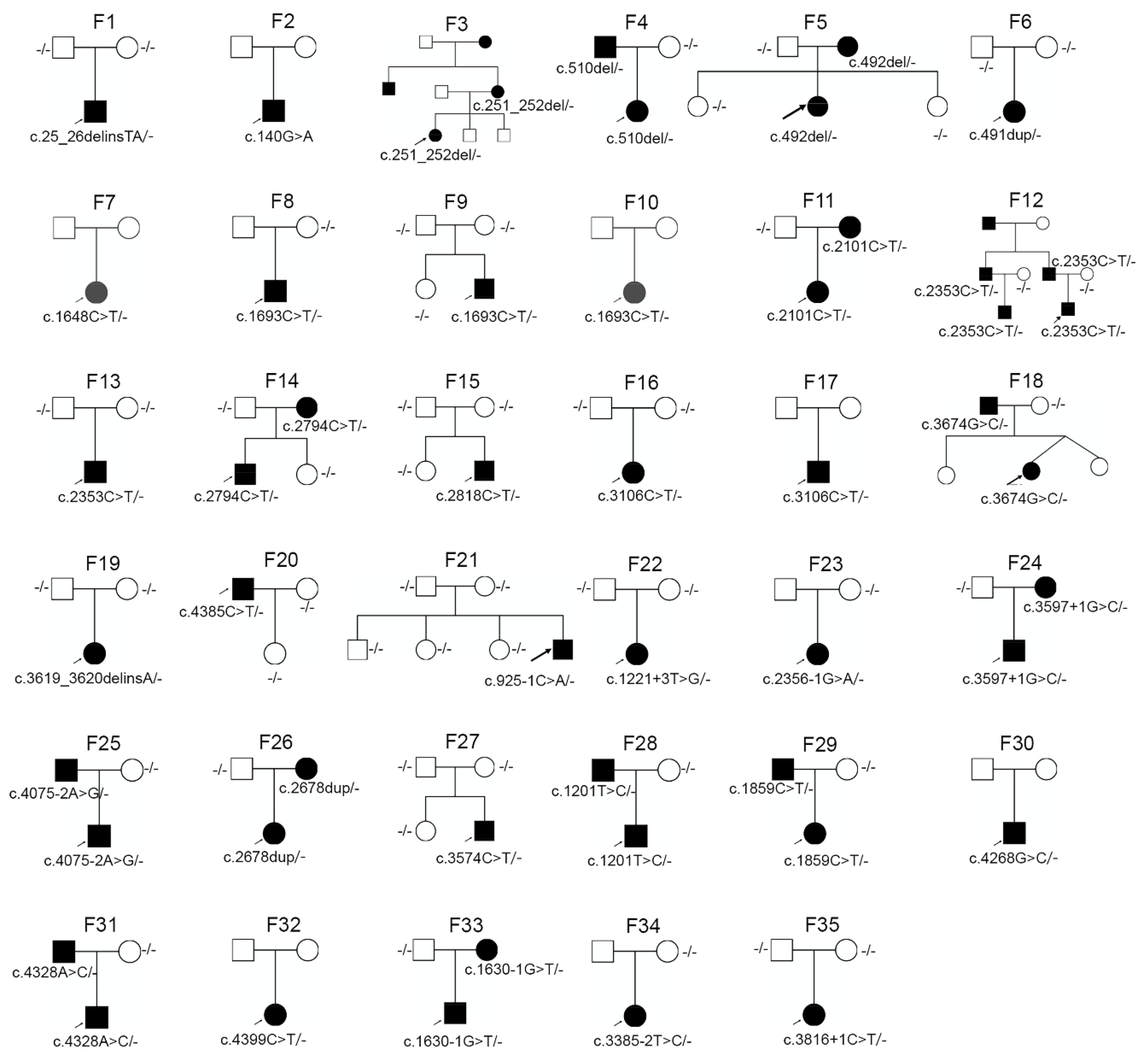
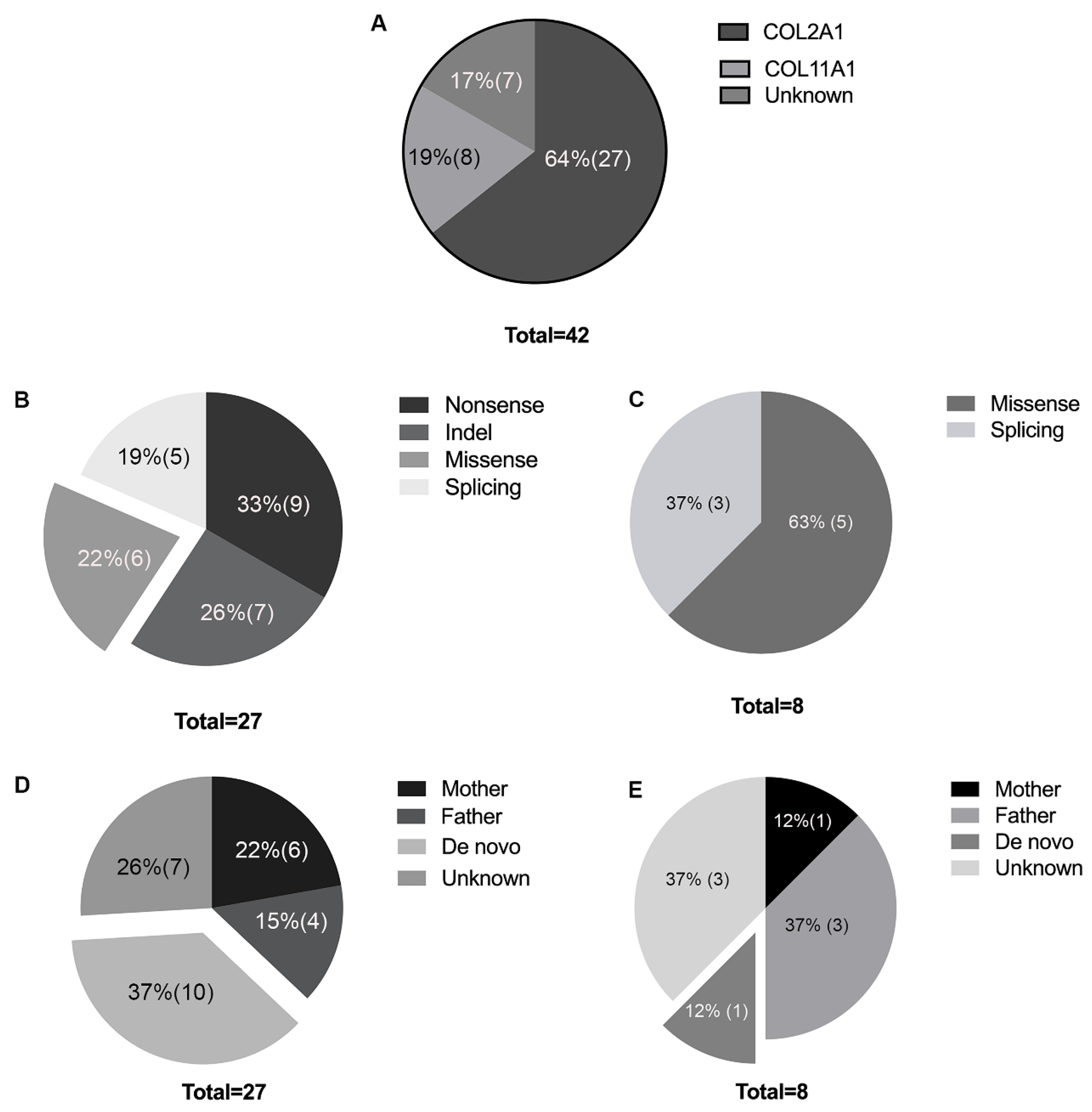
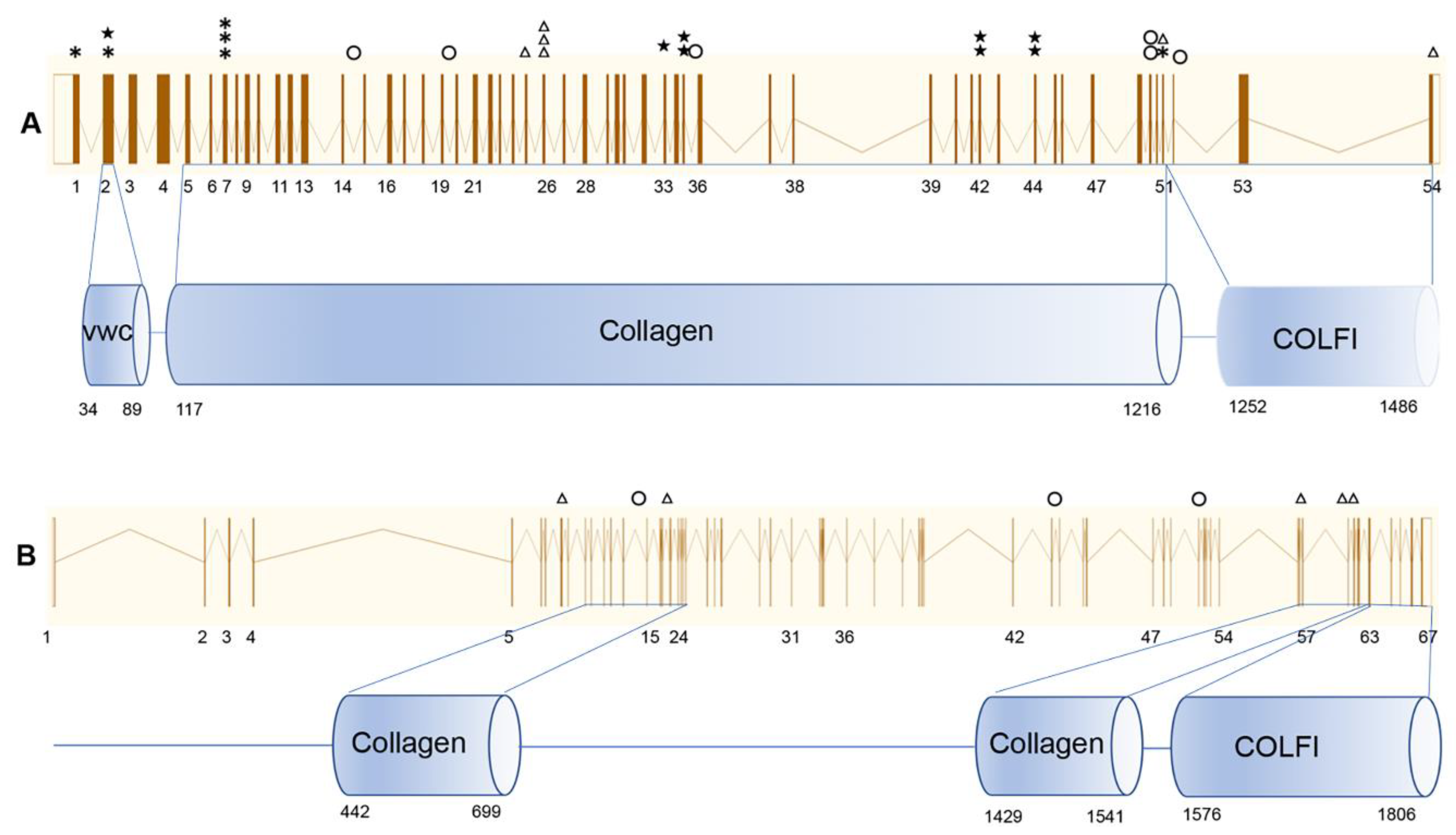
3.2. Genetic Analysis
3.3. De Novo Mutations in COL2A1 and COL11A1
3.4. Clinical Manifestation in Probands with COL2A1 Missense Mutations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stickler, G.B.; Belau, P.G.; Farrell, F.J.; Jones, J.D.; Pugh, D.G.; Steinberg, A.G.; Ward, L.E. Hereditary Progressive Arthro-Ophthalmopathy. Mayo Clin. Proc. 1965, 40, 433–455. [Google Scholar] [PubMed]
- Robin, N.H.; Moran, R.T.; Ala-Kokko, L. Stickler Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Liberfarb, R.M.; Goldblatt, A. Prevalence of mitral-valve prolapse in the Stickler syndrome. Am. J. Med. Genet. 1986, 24, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Francomano, C.A.; Liberfarb, R.M.; Hirose, T.; Maumenee, I.H.; Streeten, E.A.; Meyers, D.A.; Pyeritz, R.E. The Stickler syndrome: Evidence for close linkage to the structural gene for type II collagen. Genomics 1987, 1, 293–296. [Google Scholar] [CrossRef]
- Richards, A.J.; Yates, J.R.; Williams, R.; Payne, S.J.; Pope, F.M.; Scott, J.D.; Snead, M.P. A family with Stickler syndrome type 2 has a mutation in the COL11A1 gene resulting in the substitution of glycine 97 by valine in alpha 1 (XI) collagen. Hum. Mol. Genet. 1996, 5, 1339–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melkoniemi, M.; Brunner, H.G.; Manouvrier, S.; Hennekam, R.; Superti-Furga, A.; Kaariainen, H.; Pauli, R.M.; van Essen, T.; Warman, M.L.; Bonaventure, J.; et al. Autosomal recessive disorder otospondylomegaepiphyseal dysplasia is associated with loss-of-function mutations in the COL11A2 gene. Am. J. Hum. Genet. 2000, 66, 368–377. [Google Scholar] [CrossRef] [Green Version]
- Van Camp, G.; Snoeckx, R.L.; Hilgert, N.; van den Ende, J.; Fukuoka, H.; Wagatsuma, M.; Suzuki, H.; Smets, R.M.; Vanhoenacker, F.; Declau, F.; et al. A new autosomal recessive form of Stickler syndrome is caused by a mutation in the COL9A1 gene. Am. J. Hum. Genet. 2006, 79, 449–457. [Google Scholar] [CrossRef] [Green Version]
- Baker, S.; Booth, C.; Fillman, C.; Shapiro, M.; Blair, M.P.; Hyland, J.C.; Ala-Kokko, L. A loss of function mutation in the COL9A2 gene causes autosomal recessive Stickler syndrome. Am. J. Med. Genet. Part A 2011, 155A, 1668–1672. [Google Scholar] [CrossRef]
- Nakashima, E.; Kitoh, H.; Maeda, K.; Haga, N.; Kosaki, R.; Mabuchi, A.; Nishimura, G.; Ohashi, H.; Ikegawa, S. Novel COL9A3 mutation in a family with multiple epiphyseal dysplasia. Am. J. Med. Genet. Part A 2005, 132A, 181–184. [Google Scholar] [CrossRef]
- Alzahrani, F.; Al Hazzaa, S.A.; Tayeb, H.; Alkuraya, F.S. LOXL3, encoding lysyl oxidase-like 3, is mutated in a family with autosomal recessive Stickler syndrome. Hum. Genet. 2015, 134, 451–453. [Google Scholar] [CrossRef]
- Snead, M.P.; Yates, J.R. Clinical and Molecular genetics of Stickler syndrome. J. Med. Genet. 1999, 36, 353–359. [Google Scholar]
- Hoornaert, K.P.; Vereecke, I.; Dewinter, C.; Rosenberg, T.; Beemer, F.A.; Leroy, J.G.; Bendix, L.; Bjorck, E.; Bonduelle, M.; Boute, O.; et al. Stickler syndrome caused by COL2A1 mutations: Genotype-phenotype correlation in a series of 100 patients. Eur. J. Hum. Genet. EJHG 2010, 18, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Ding, X.; Li, J.; Hu, A.; Yuan, M.; Yang, Y.; Zhan, Z.; Li, Z.; Lu, L. Novel mutations in FZD4 and phenotype-genotype correlation in Chinese patients with familial exudative vitreoretinopathy. Mol. Vis. 2016, 22, 917–932. [Google Scholar] [PubMed]
- Barat-Houari, M.; Dumont, B.; Fabre, A.; Them, F.T.; Alembik, Y.; Alessandri, J.L.; Amiel, J.; Audebert, S.; Baumann-Morel, C.; Blanchet, P.; et al. The expanding spectrum of COL2A1 gene variants IN 136 patients with a skeletal dysplasia phenotype. Eur. J. Hum. Genet. EJHG 2016, 24, 992–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Huang, L.; Xu, Y.; Xiao, X.; Li, S.; Jia, X.; Gao, B.; Wang, P.; Guo, X.; Zhang, Q. Exome Sequencing on 298 Probands With Early-Onset High Myopia: Approximately One-Fourth Show Potential Pathogenic Mutations in RetNet Genes. Investig. Ophthalmol. Vis. Sci. 2015, 56, 8365–8372. [Google Scholar] [CrossRef] [Green Version]
- Richards, A.J.; Laidlaw, M.; Whittaker, J.; Treacy, B.; Rai, H.; Bearcroft, P.; Baguley, D.M.; Poulson, A.; Ang, A.; Scott, J.D.; et al. High efficiency of mutation detection in type 1 stickler syndrome using a two-stage approach: Vitreoretinal assessment coupled with exon sequencing for screening COL2A1. Hum. Mutat. 2006, 27, 696–704. [Google Scholar] [CrossRef]
- Savasta, S.; Salpietro, V.; Sparta, M.V.; Foiadelli, T.; Laino, D.; Lobefalo, L.; Marseglia, G.L.; Verrotti, A. Stickler syndrome associated with epilepsy: Report of three cases. Eur. J. Pediatr. 2015, 174, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Annunen, S.; Korkko, J.; Czarny, M.; Warman, M.L.; Brunner, H.G.; Kaariainen, H.; Mulliken, J.B.; Tranebjaerg, L.; Brooks, D.G.; Cox, G.F.; et al. Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am. J. Hum. Genet. 1999, 65, 974–983. [Google Scholar] [CrossRef] [Green Version]
- Kondo, H.; Matsushita, I.; Nagata, T.; Hayashi, T.; Kakinoki, M.; Uchio, E.; Kondo, M.; Ohji, M.; Kusaka, S. Novel mutations in the COL2A1 gene in Japanese patients with Stickler syndrome. Hum. Genome. Var. 2016, 3, 16018. [Google Scholar] [CrossRef]
- Zechi-Ceide, R.M.; Jesus Oliveira, N.A.; Guion-Almeida, M.L.; Antunes, L.F.; Richieri-Costa, A.; Passos-Bueno, M.R. Clinical evaluation and COL2A1 gene analysis in 21 Brazilian families with Stickler syndrome: Identification of novel mutations, further genotype/phenotype correlation, and its implications for the diagnosis. Eur. J. Med. Genet. 2008, 51, 183–196. [Google Scholar] [CrossRef]
- Richards, A.J.; Martin, S.; Yates, J.R.; Scott, J.D.; Baguley, D.M.; Pope, F.M.; Snead, M.P. COL2A1 exon 2 mutations: Relevance to the Stickler and Wagner syndromes. Br. J. Ophthalmol. 2000, 84, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Ala-Kokko, L.; Shanske, A.L. Mosaicism in Marshall Syndrome. Am. J. Med. Genet. Part A 2009, 149, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Acke, F.R.; Malfait, F.; Vanakker, O.M.; Steyaert, W.; De Leeneer, K.; Mortier, G.; Dhooge, I.; De Paepe, A.; De Leenheer, E.M.; Coucke, P.J. Novel pathogenic COL11A1/COL11A2 variants in Stickler syndrome detected by targeted NGS and exome sequencing. Mol. Genet. Metab. 2014, 113, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Nikopoulos, K.; Schrauwen, I.; Simon, M.; Collin, R.W.; Veckeneer, M.; Keymolen, K.; Van Camp, G.; Cremers, F.P.; van den Born, L.I. Autosomal recessive Stickler syndrome in two families is caused by mutations in the COL9A1 gene. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4774–4779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasa, Y.; Moteki, H.; Hattori, M.; Sato, R.; Nishio, S.Y.; Takumi, Y.; Usami, S. Non-ocular Stickler syndrome with a novel mutation in COL11A2 diagnosed by massively parallel sequencing in Japanese hearing loss patients. Ann. Otol. Rhinol. Laryngol. 2015, 124 (Suppl. 1), 111S–117S. [Google Scholar] [CrossRef]
- Faletra, F.; D’Adamo, A.P.; Bruno, I.; Athanasakis, E.; Biskup, S.; Esposito, L.; Gasparini, P. Autosomal recessive Stickler syndrome due to a loss of function mutation in the COL9A3 gene. Am. J. Med. Genet. Part A 2014, 164A, 42–47. [Google Scholar] [CrossRef]
- Admiraal, R.J.; Brunner, H.G.; Dijkstra, T.L.; Huygen, P.L.; Cremers, C.W. Hearing loss in the nonocular Stickler syndrome caused by a COL11A2 mutation. Laryngoscope 2000, 110, 457–461. [Google Scholar] [CrossRef]
- Nixon, T.R.W.; Alexander, P.; Richards, A.; McNinch, A.; Bearcroft, P.W.P.; Cobben, J.; Snead, M.P. Homozygous Type IX collagen variants (COL9A1, COL9A2, and COL9A3) causing recessive Stickler syndrome-Expanding the phenotype. Am. J. Med. Genet. Part A 2019, 179, 1498–1506. [Google Scholar] [CrossRef]
- Spickett, C.; Hysi, P.; Hammond, C.J.; Prescott, A.; Fincham, G.S.; Poulson, A.V.; McNinch, A.M.; Richards, A.J.; Snead, M.P. Deep Intronic Sequence Variants in COL2A1 Affect the Alternative Splicing Efficiency of Exon 2, and May Confer a Risk for Rhegmatogenous Retinal Detachment. Hum. Mutat. 2016, 37, 1085–1096. [Google Scholar] [CrossRef] [Green Version]
- Acuna-Hidalgo, R.; Veltman, J.A.; Hoischen, A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 2016, 17, 241. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.B.; Bigham, A.W.; Buckingham, K.J.; Hannibal, M.C.; McMillin, M.J.; Gildersleeve, H.I.; Beck, A.E.; Tabor, H.K.; Cooper, G.M.; Mefford, H.C.; et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010, 42, 790–793. [Google Scholar] [CrossRef] [Green Version]
- de Ligt, J.; Willemsen, M.H.; van Bon, B.W.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; de Vries, P.; Gilissen, C.; et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldmann, J.M.; Wong, W.S.; Pinelli, M.; Farrah, T.; Bodian, D.; Stittrich, A.B.; Glusman, G.; Vissers, L.E.; Hoischen, A.; Roach, J.C.; et al. Parent-of-origin-specific signatures of de novo mutations. Nat. Genet. 2016, 48, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Kong, A.; Frigge, M.L.; Masson, G.; Besenbacher, S.; Sulem, P.; Magnusson, G.; Gudjonsson, S.A.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 2012, 488, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Total | COL2A1 | COL11A1 | Unknown |
|---|---|---|---|---|
| Number (total) | 42 | 27 (64%) | 8 (19%) | 7 (21%) |
| Male (n, %) | 23 (55%) | 14 (52%) | 4 (50%) | 5 (71%) |
| Female (n, %) | 19 (45%) | 13 (48%) | 4 (50%) | 2 (29%) |
| Age (years) (median (P25, P75)) | 8 (5, 10.25) | 7 (5, 11) | 10 (5.75, 20.25) | 12 (4, 18) |
| AL (mm) (median (P25, P75)) | 27.20 (24.15, 29.04) | 27.06 (24.99, 28.59) | 24.96 (23.58, 29.16) | 28.71 (21.30, 29.83) |
| SE (D) (Mean ± SD) | −11.91 ± 5.37 | −11.64 ± 4.95 | −9.43 ± 7.27 | −15.58 ± 3.74 |
| HM (n, %) | 32 (76%) | 21 (78%) | 5 (63%) | 6 (86%) |
| RD (n, %) | 29 (69%) | 20 (74%) | 4 (50%) | 5 (71%) |
| RD (eyes, %) | 37 (44%) | 25 (46%) | 5 (31%) | 7 (50%) |
| Glaucoma (n, %) | 5 (12%) | 5 (19%) | 0 | 0 |
| Cataract (n, %) | 10 (24%) | 6 (22%) | 2 (25%) | 2 (29%) |
| Family ID | Sequence | Gene | Exon | Mutation Type | Location | DNA Change | Amino Acid Change | ExAC—All | ExAC—East Asian | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | TGP | COL2A1 | 1 | indel | chr12: 48398079 | c.25_26delinsTA | p.Thr9* | 0 | 0 | novel |
| 2 | TGP | COL2A1 | 2 | nonsense | chr12:48393854 | c.140G > A | p.Trp47* | 0 | 0 | novel |
| 3 | TGP | COL2A1 | 2 | indel | chr12:48393736 | c.251_252del | p.Glu84Valfs*8 | 0 | 0 | novel |
| 4 | TGP | COL2A1 | 7 | indel | chr12:48391409 | c.510del | p.Gly171Valfs*28 | 0 | 0 | [21] |
| 5 | WES | COL2A1 | 7 | indel | chr12:48393736 | c.492del | p.Gly165Valfs*34 | 0 | 0 | novel |
| 6 | TGP | COL2A1 | 7 | indel | chr12:48391428 | c.491dup | p.Gly165Trpfs*24 | 0 | 0 | novel |
| 7 | TGP | COL2A1 | 25 | missense | chr12:48379543 | c.1648C > T | p.Arg550Cys | 0 | 0 | [14] |
| 8 | WES | COL2A1 | 26 | missense | chr12:48379358 | c.1693C > T | p.Arg565Cys | 0 | 0 | [15] |
| 9 | TGP | COL2A1 | 26 | missense | chr12:48379358 | c.1693C > T | p.Arg565Cys | 0 | 0 | [15] |
| 10 | TGP | COL2A1 | 26 | missense | chr12:48379358 | c.1693C > T | p.Arg565Cys | 0 | 0 | [15] |
| 11 | WES | COL2A1 | 33 | nonsense | chr12:48376723 | c.2101C > T | p.Arg701* | 0 | 0 | [16] |
| 12 | TGP | COL2A1 | 35 | nonsense | chr12:48375892 | c.2353C > T | p.Arg785* | 0 | 0 | [21] |
| 13 | WES | COL2A1 | 35 | nonsense | chr12:48375892 | c.2353C > T | p.Arg785* | 0 | 0 | [21] |
| 14 | TGP | COL2A1 | 42 | nonsense | chr12:48372481 | c.2794C > T | p.Arg932* | 0 | 0 | [15] |
| 15 | WES | COL2A1 | 42 | nonsense | chr12:48372457 | c.2818C > T | p.Arg940* | 0 | 0 | [19] |
| 16 | TGP | COL2A1 | 44 | nonsense | chr12:48371798 | c.3106C > T | p.Arg1036* | 1/114802 | 0/8542 | [17] |
| 17 | TGP | COL2A1 | 44 | nonsense | chr12:48371798 | c.3106C > T | p.Arg1036* | 1/114802 | 0/8542 | [17] |
| 18 | TGP | COL2A1 | 51 | missense | chr12:48369312 | c.3674G > C | p.Gly1225Ala | 0 | 0 | novel |
| 19 | WES | COL2A1 | 51 | indel | chr12:48369362 | c.3619_3620delinsA | p.Pro1207Thrfs*20 | 0 | 0 | novel |
| 20 | TGP | COL2A1 | 54 | missense | chr12:48367279 | c.4385C > T | p.Arg1459Cys | 3/121410 | 0/8654 | novel |
| 21 | WES | COL2A1 | Intro 14 | splicing | chr12:48387286 | c.925-1C > A | _ | 0 | 0 | [18] |
| 22 | WES | COL2A1 | Intro 19 | splicing | chr12:48381391 | c.1221+3T > G | _ | 0 | 0 | novel |
| 23 | TGP | COL2A1 | Intro 35 | splicing | chr12:48375613 | c.2356-1G > A | 0 | 0 | novel | |
| 24 | TGP | COL2A1 | Intro 50 | splicing | chr12:48369745 | c.3597+1G > C | _ | 0 | 0 | novel |
| 25 | WES | COL2A1 | Intro 52 | splicing | chr12:48368116 | c.4075-2A > G | 0 | 0 | Novel | |
| 26 | WES | COL2A1 | 40 | indel | chr12:48373792 | c.2678dup | p.Pro893fs | 0 | 0 | [12] |
| 27 | WES | COL2A1 | 50 | nonsense | chr12:48369769 | c.3574C > T | p.Arg1192* | 0 | 0 | [20] |
| 28 | TGP | COL11A1 | 8 | missense | chr1:103488342 | c.1201T > C | p.Phe401Leu | 1/119952 | 1/8594 | novel |
| 29 | WES | COL11A1 | 19 | missense | chr1:103470204 | c.1859C > T | p.Pro620Leu | 0 | 0 | novel |
| 30 | WES | COL11A1 | 57 | missense | chr1:103363724 | c.4268G > C | p.Gly1423Ala | 0 | 0 | novel |
| 31 | WES | COL11A1 | 58 | missense | chr1:103356035 | c.4328A > C | p.Lys1443Thr | 4/102722 | 2/7546 | novel |
| 32 | TGP | COL11A1 | 59 | missense | chr1:103355112 | c.4399C > T | p.Gln1467* | 2/120730 | 2/8600 | novel |
| 33 | WES | COL11A1 | intro 14 | splicing | chr1:103474073 | c.1630-1G > T | 0 | 0 | novel | |
| 34 | TGP | COL11A1 | intro 43 | splicing | chr1:103404646 | c.3385-2T > C | _ | 0 | 0 | [18] |
| 35 | TGP | COL11A1 | intro 50 | splicing | chr1:103381186 | c.3816+1C > T | _ | 0 | 0 | [22] |
| Family ID | Gene | DNA Changes | Amino Acid Changes | Paternal Childbearing Age (Years) | Maternal Childbearing Age (Years) |
|---|---|---|---|---|---|
| 1 | COL2A1 | c.25_26delinsTA | p.Thr9* | 30 | 31 |
| 6 | COL2A1 | c.491dup | p.Gly165Trpfs*24 | 30 | 30 |
| 9 | COL2A1 | c.1693C > T | p.Arg565Cys | 29 | 21 |
| 13 | COL2A1 | c.2353C > T | p.Arg785* | 24 | 23 |
| 15 | COL2A1 | c.2818C > T | p.Arg940* | 34 | 33 |
| 16 | COL2A1 | c.3106C > T | p.Arg1036* | 26 | 23 |
| 19 | COL2A1 | c.3619_3620delinsA | p.Pro1207Thrfs*20 | 33 | 33 |
| 21 | COL2A1 | c.925-1C > A | _ | 38 | 28 |
| 22 | COL2A1 | c.1221 + 3T > G | _ | 34 | 30 |
| 27 | COL2A1 | c.3574C > T | p.R1192* | 21 | 19 |
| 35 | COL11A1 | c.3816 + 1C > T | _ | 27 | 30 |
| Mean ± SD | 29.64 ± 4.97 | 27.36 ± 4.97 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, L.; Chen, C.; Wang, Z.; Sun, L.; Li, S.; Zhang, T.; Luo, X.; Ding, X. Mutation Spectrum and De Novo Mutation Analysis in Stickler Syndrome Patients with High Myopia or Retinal Detachment. Genes 2020, 11, 882. https://doi.org/10.3390/genes11080882
Huang L, Chen C, Wang Z, Sun L, Li S, Zhang T, Luo X, Ding X. Mutation Spectrum and De Novo Mutation Analysis in Stickler Syndrome Patients with High Myopia or Retinal Detachment. Genes. 2020; 11(8):882. https://doi.org/10.3390/genes11080882
Chicago/Turabian StyleHuang, Li, Chonglin Chen, Zhirong Wang, Limei Sun, Songshan Li, Ting Zhang, Xiaoling Luo, and Xiaoyan Ding. 2020. "Mutation Spectrum and De Novo Mutation Analysis in Stickler Syndrome Patients with High Myopia or Retinal Detachment" Genes 11, no. 8: 882. https://doi.org/10.3390/genes11080882
APA StyleHuang, L., Chen, C., Wang, Z., Sun, L., Li, S., Zhang, T., Luo, X., & Ding, X. (2020). Mutation Spectrum and De Novo Mutation Analysis in Stickler Syndrome Patients with High Myopia or Retinal Detachment. Genes, 11(8), 882. https://doi.org/10.3390/genes11080882