Unveiling Sex-Based Differences in the Effects of Alcohol Abuse: A Comprehensive Functional Meta-Analysis of Transcriptomic Studies


Abstract
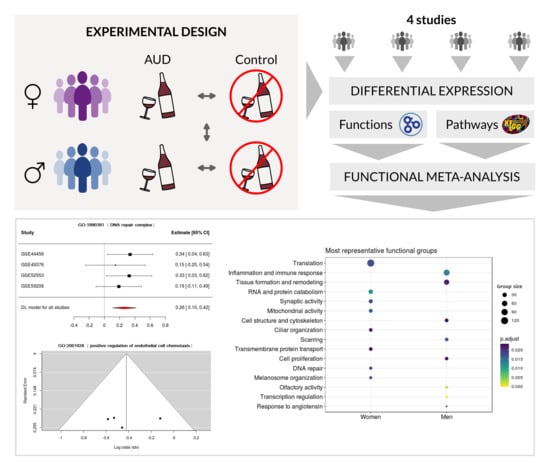
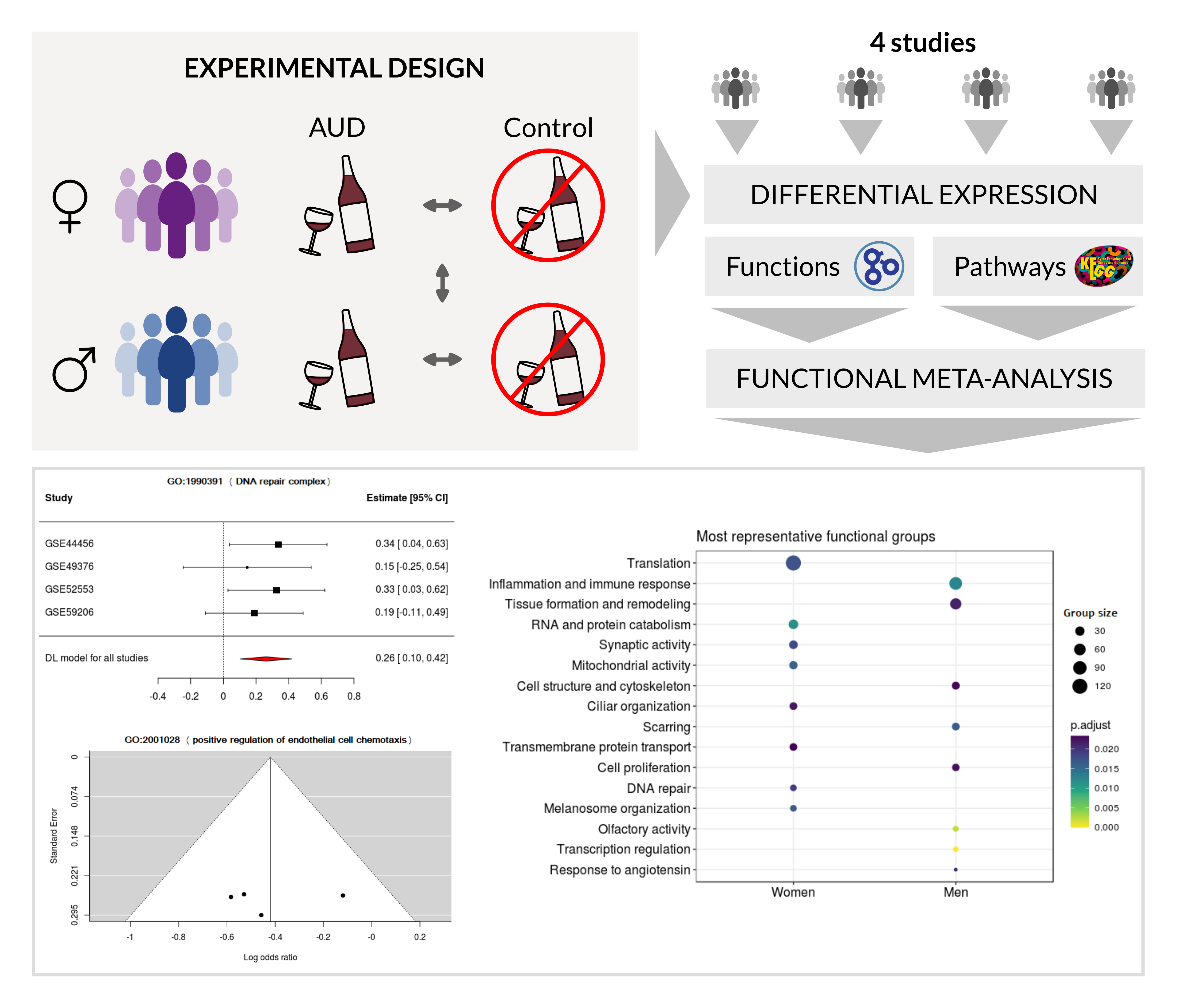
:
1. Introduction
2. Materials and Methods
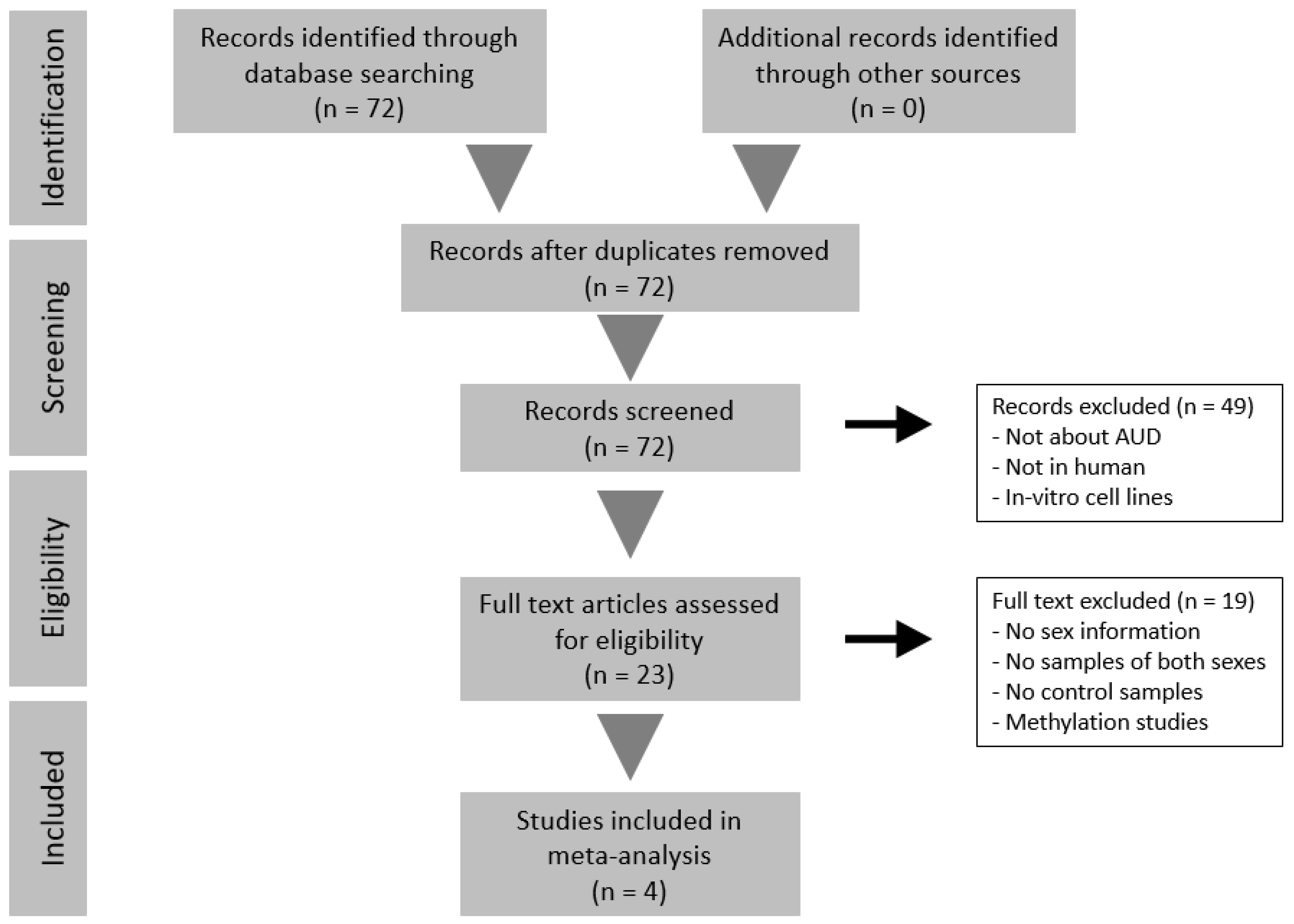
2.1. Systematic Review and Study Selection
2.2. Bioinformatics Analysis Strategy
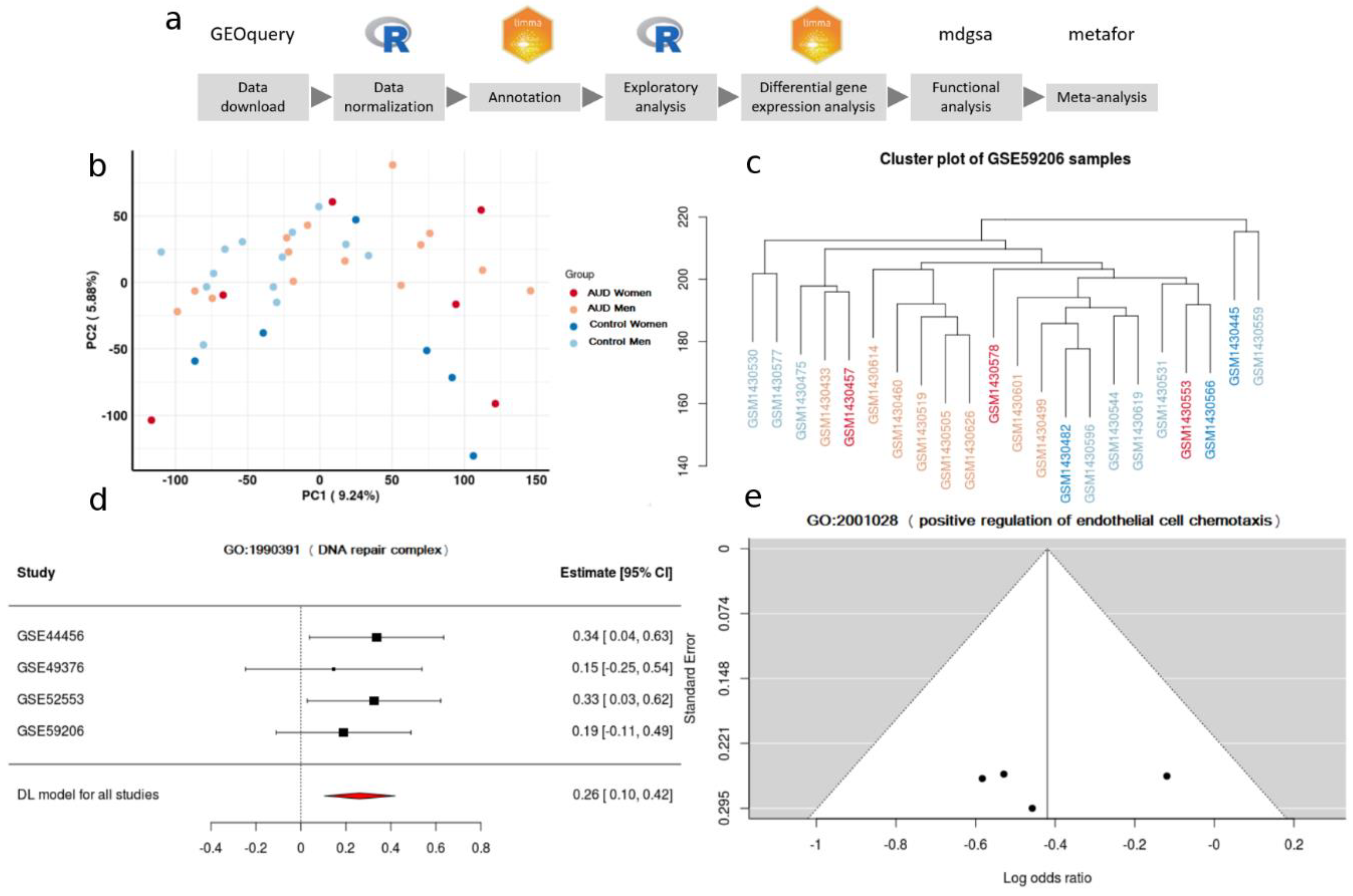
2.3. Data Processing and Exploratory Analysis
2.4. Differential Expression Analysis and Functional Profiling
2.5. Meta-Analysis
2.6. Web Tools
3. Results
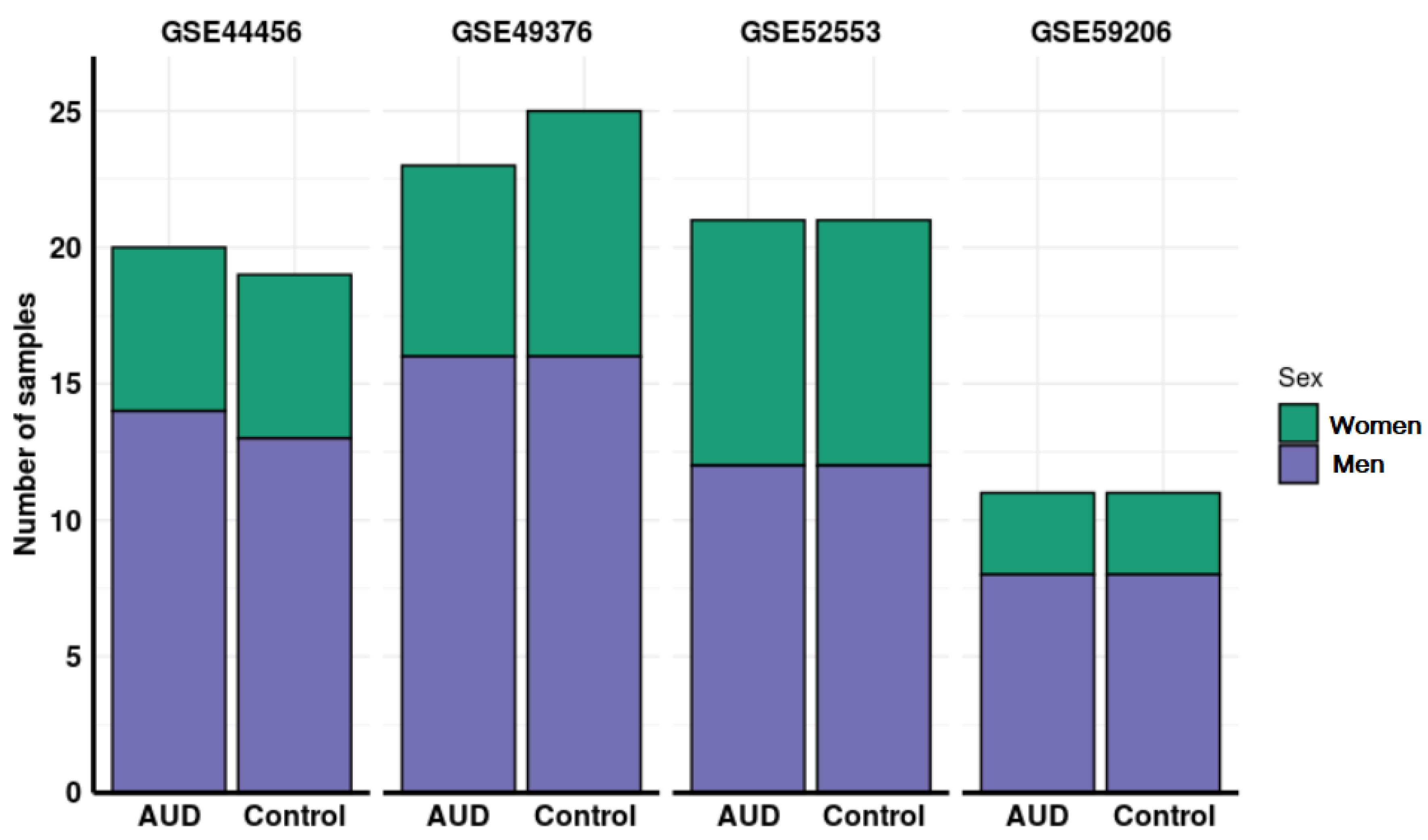
3.1. Systematic Review and Study Selection
3.2. Individual Analysis of the Studies
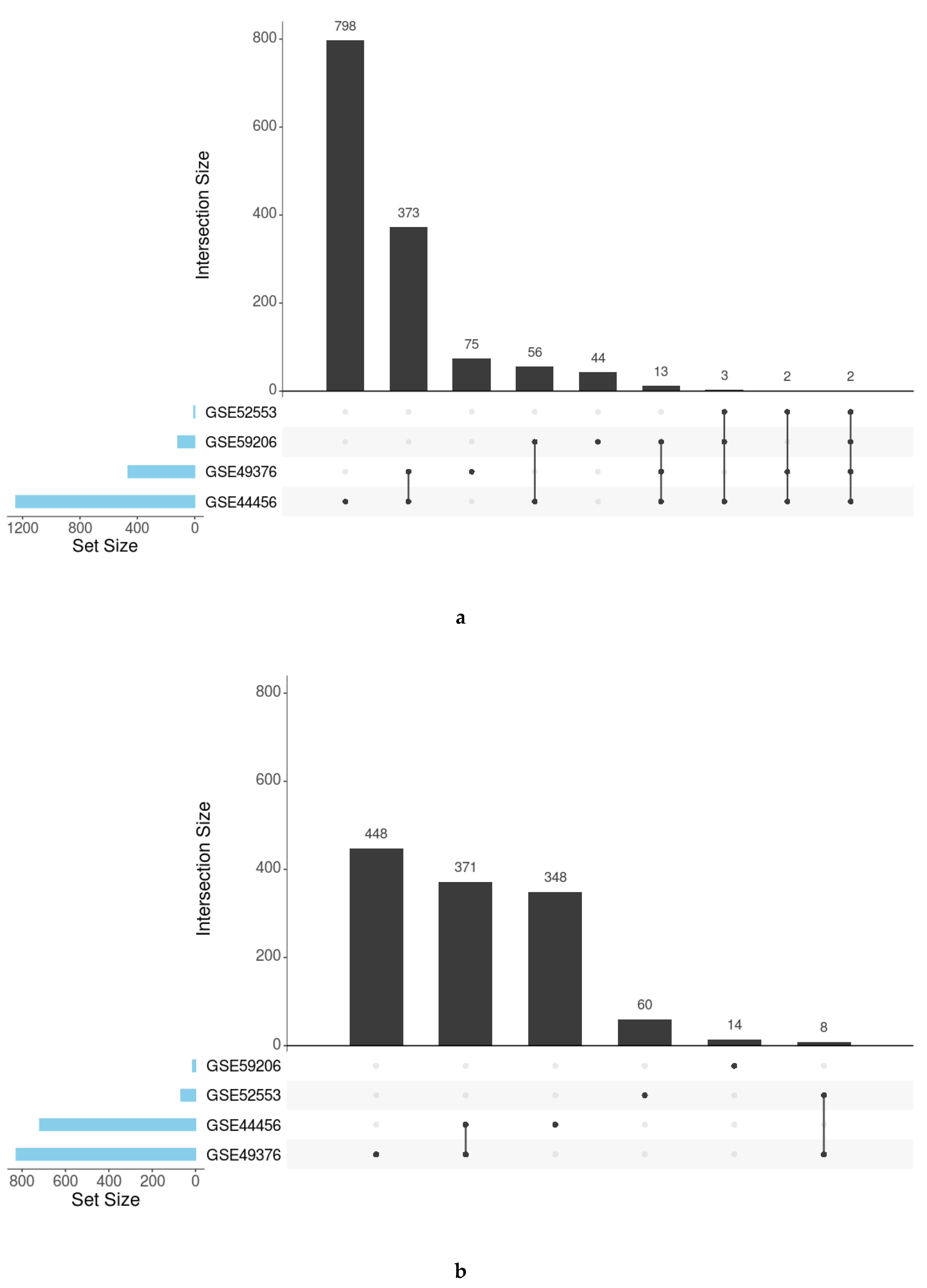
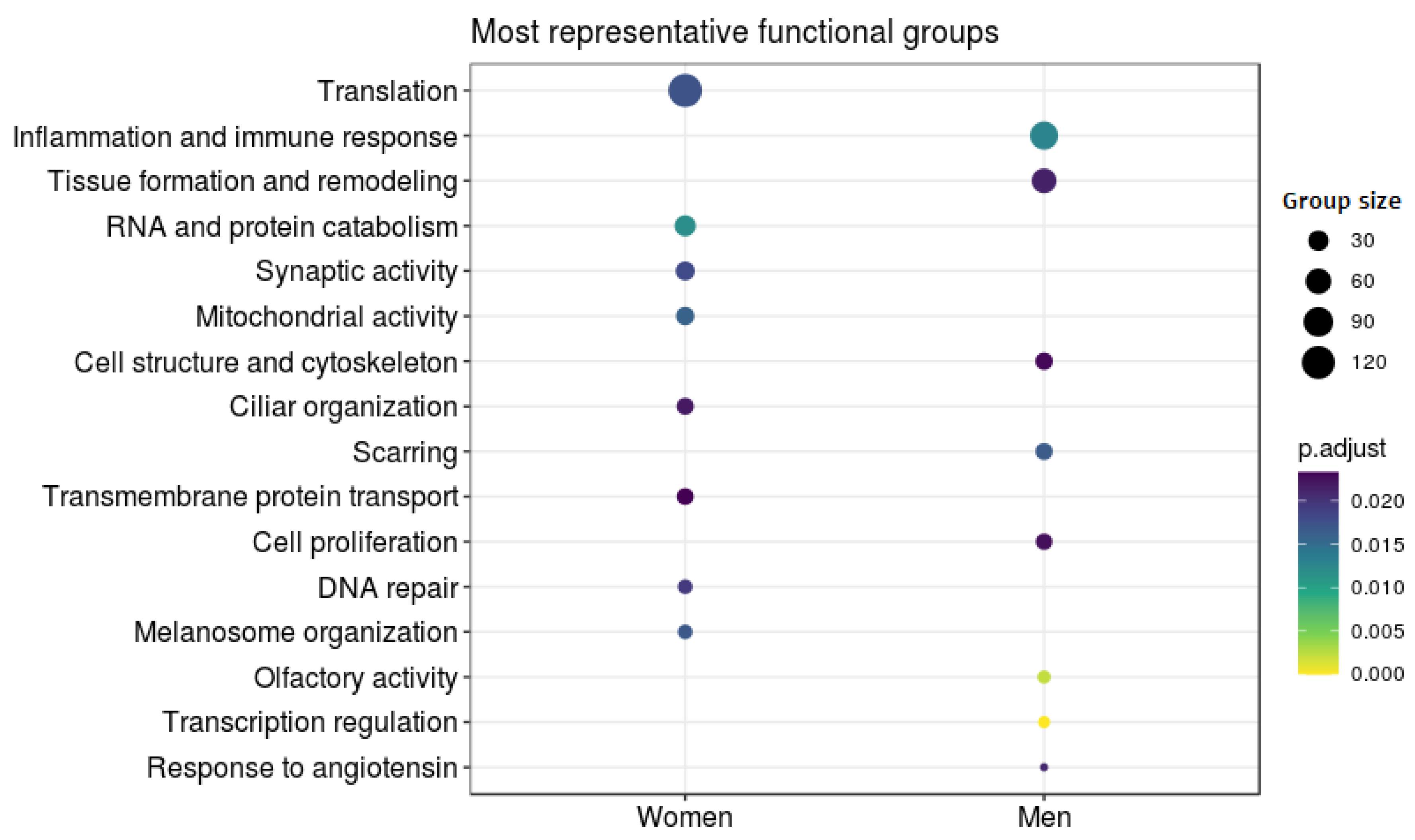
3.3. Meta-Analysis
3.4. Metafun-AUD Web Tool
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- World Health Organization. Global Status Report on Alcohol and Health 2018; Poznyak, V., Rekve, D., Eds.; World Health Organization: Geneva, Switzerland, 2018; ISBN 978-92-4-156563-9. [Google Scholar]
- World Health Organization. International Statistical Classification of Diseases and Related Health Problems, 11th Revision; World Health Organization: Geneva, Switzerland, 2018; Volume 6, Available online: https://icd.who.int/browse11/l-m/en2018 (accessed on 5 May 2020).
- Enoch, M.-A. Genetic and Environmental Influences on the Development of Alcoholism: Resilience vs. Risk. Ann. N. Y. Acad. Sci. 2006, 1094, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Wilsnack, S.C.; Vogeltanz, N.D.; Klassen, A.D.; Harris, T.R. Childhood sexual abuse and women’s substance abuse: National survey findings. J. Stud. Alcohol 1997, 58, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Edenberg, H.J. The Genetics of Alcohol Metabolism: Role of Alcohol Dehydrogenase and Aldehyde Dehydrogenase Variants. Alcohol Res. Heal. J. Natl. Inst. Alcohol Abus. Alcohol. 2007, 30, 5–13. [Google Scholar]
- Chen, C.-H.; Ferreira, J.C.; Gross, E.R.; Mochly-Rosen, D. Targeting aldehyde dehydrogenase 2: New therapeutic opportunities. Physiol. Rev. 2014, 94, 1–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumark, Y.D.; Friedlander, Y.; Thomasson, H.R.; Li, T.-K. Association of the ADH2*2 allele with reduced ethanol consumption in Jewish men in Israel: A pilot study. J. Stud. Alcohol 1998, 59, 133–139. [Google Scholar] [CrossRef]
- Pascual, M.; Baliño, P.; Alfonso-Loeches, S.; Aragon, C.M.; Guerri, C. Impact of TLR4 on behavioral and cognitive dysfunctions associated with alcohol-induced neuroinflammatory damage. Brain Behav. Immun. 2011, 25, S80–S91. [Google Scholar] [CrossRef]
- Witt, E.D. Puberty, hormones, and sex differences in alcohol abuse and dependence. Neurotoxicol. Teratol. 2007, 29, 81–95. [Google Scholar] [CrossRef] [Green Version]
- Garfinkel, L.; Boffetta, P.; Stellman, S.D. Alcohol and breast cancer: A cohort study. Prev. Med. 1988, 17, 686–693. [Google Scholar] [CrossRef] [Green Version]
- Manzoni, C.; A Kia, D.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; A Lewis, P.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Briefings Bioinform. 2016, 19, 286–302. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- García-García, F. Methods of Functional Enrichment Analysis in Genomic Studies. Ph.D. Thesis, University of Valencia, Valencia, Spain, 2016. [Google Scholar]
- Normand, S.L.T. Meta-analysis: Formulating, evaluating, combining, and reporting. Stat. Med. 1999, 18, 248–287. [Google Scholar] [CrossRef]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.A.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA Statement for Reporting Systematic Reviews and Meta-Analyses of Studies That Evaluate Health Care Interventions: Explanation and Elaboration. PLoS Med. 2009, 6, e1000100. [Google Scholar] [CrossRef] [PubMed]
- Voigt, A.P.; Mulfaul, K.; Mullin, N.K.; Flamme-Wiese, M.J.; Giacalone, J.C.; Stone, E.M.; Tucker, B.A.; Scheetz, T.E.; Mullins, R.F. Single-cell transcriptomics of the human retinal pigment epithelium and choroid in health and macular degeneration. Proc. Natl. Acad. Sci. USA 2019, 116, 24100–24107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolesnikov, N.; Hastings, E.; Keays, M.; Melnichuk, O.; Tang, Y.; Williams, E.; Dylag, M.; Kurbatova, N.; Brandizi, M.; Burdett, T.; et al. ArrayExpress update—simplifying data submissions. Nucleic Acids Res. 2014, 43, D1113–D1116. [Google Scholar] [CrossRef]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Maglott, D.; Ostell, J.; Pruitt, K.D.; Tatusova, T. Entrez Gene: Gene-centered information at NCBI. Nucleic Acids Res. 2010, 39, D52–D57. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Montaner, D.; Dopazo, J. Multidimensional Gene Set Analysis of Genomic Data. PLoS ONE 2010, 5, e10348. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2018, 47, D590–D595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2016, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2018, 47, D330–D338. [Google Scholar] [CrossRef] [Green Version]
- Lex, A.; Gehlenborg, N. Sets and intersections. Nat. Methods 2014, 11, 779. [Google Scholar] [CrossRef]
- Viechtbauer, W. Conducting Meta-Analyses inRwith themetaforPackage. J. Stat. Softw. 2010, 36, 1–48. [Google Scholar] [CrossRef] [Green Version]
- DerSimonian, R.; Laird, N. Meta-analysis in clinical trials. Control. Clin. Trials 1986, 7, 177–188. [Google Scholar] [CrossRef]
- McClintick, J.N.; Xuei, X.; Tischfield, J.; Goate, A.; Foroud, T.; Wetherill, L.; Ehringer, M.A.; Edenberg, H.J. Stress–response pathways are altered in the hippocampus of chronic alcoholics. Alcohol 2013, 47, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Wang, F.; Liu, Y.; Yu, Y.; Gelernter, J.; Zhang, H. Sex-biased methylome and transcriptome in human prefrontal cortex. Hum. Mol. Genet. 2013, 23, 1260–1270. [Google Scholar] [CrossRef] [Green Version]
- Sheedy, D.; Garrick, T.; Dedova, I.; Hunt, C.; Miller, R.; Sundqvist, N.; Harper, C. An Australian Brain Bank: A critical investment with a high return! Cell Tissue Bank. 2008, 9, 205–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castle, D.J.; Jablensky, A.; McGrath, J.J.; Carr, V.; Morgan, V.; Waterreus, A.; Valuri, G.; Stain, H.; McGuffin, P.; Farmer, A. The diagnostic interview for psychoses (DIP): Development, reliability and applications. Psychol. Med. 2005, 36, 69–80. [Google Scholar] [CrossRef] [PubMed]
- McClintick, J.N.; Brooks, A.I.; Deng, L.; Liang, L.; Wang, J.C.; Kapoor, M.; Xuei, X.; Foroud, T.; Tischfield, J.; Edenberg, H.J. Ethanol treatment of lymphoblastoid cell lines from alcoholics and non-alcoholics causes many subtle changes in gene expression. Alcohology 2014, 48, 603–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foroud, T.; Edenberg, H.J.; Goate, A.; Rice, J.; Flury, L.; Koller, D.L.; Bierut, L.J.; Conneally, P.M.; Nurnberger, J.I.; Bucholz, K.K.; et al. Alcoholism susceptibility loci: Confirmation studies in a replicate sample and further mapping. Alcohol. Clin. Exp. Res. 2000, 24, 933–945. [Google Scholar] [CrossRef]
- Beech, R.D.; Leffert, J.J.; Lin, A.; Hong, K.A.; Hansen, J.; Umlauf, S.; Mane, S.; Zhao, H.; Sinha, R. Stress-Related Alcohol Consumption in Heavy Drinkers Correlates with Expression of miR-10a, miR-21, and Components of the TAR-RNA-Binding Protein-Associated Complex. Alcohol. Clin. Exp. Res. 2014, 38, 2743–2753. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders; American Psychiatric Association Publishing: Washington, DC, USA, 2013. [Google Scholar]
- Kim, A.M.; Tingen, C.M.; Woodruff, T.K. Sex bias in trials and treatment must end. Nature 2010, 465, 688–689. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, J.M.; Baak, A.; Blomberg, N.; Boiten, J.-W.; Santos, L.O.B.D.S.; E Bourne, P.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef] [Green Version]
- European Commission. Guidelines on FAIR Data Management in Horizon 2020. Tech. Rep. 2016. Available online: https://ec.europa.eu/research/participants/data/ref/h2020/grants_manual/hi/oa_pilot/h2020-hi-oa-data-mgt_en.pdf (accessed on 13 May 2020).
- Lewohl, J.M.; Wang, L.; Miles, M.F.; Zhang, L.; Dodd, P.R.; Harris, R.A. Gene expression in human alcoholism: Microarray analysis of frontal cortex. Alcohol. Clin. Exp. Res. 2000, 24, 1873–1882. [Google Scholar] [CrossRef]
- Liu, J.; Lewohl, J.M.; Harris, R.A.; Iyer, V.R.; Dodd, P.R.; Randall, P.K.; Mayfield, R.D. Patterns of Gene Expression in the Frontal Cortex Discriminate Alcoholic from Nonalcoholic Individuals. Neuropsychopharmacology 2005, 31, 1574–1582. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, M.; Wang, J.-C.; Farris, S.P.; Liu, Y.; McClintick, J.N.; Gupta, I.; Meyers, J.L.; Bertelsen, S.; Chao, M.; Nurnberger, J.; et al. Analysis of whole genome-transcriptomic organization in brain to identify genes associated with alcoholism. Transl. Psychiatry 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, F.; Xu, H.; Liu, Y.; Liu, J.; Zhao, H.; Gelernter, J. Differentially co-expressed genes in postmortem prefrontal cortex of individuals with alcohol use disorders: Influence on alcohol metabolism-related pathways. Qual. Life Res. 2014, 133, 1383–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Monte, S.M.; Kril, J.J. Human alcohol-related neuropathology. Acta Neuropathol. 2013, 127, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Pfefferbaum, A.; Rosenbloom, M.; Rohlfing, T.; Sullivan, E.V. Degradation of Association and Projection White Matter Systems in Alcoholism Detected with Quantitative Fiber Tracking. Boil. Psychiatry 2009, 65, 680–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crews, F.T.; Nixon, K. Mechanisms of Neurodegeneration and Regeneration in Alcoholism. Alcohol Alcohol. 2009, 44, 115–127. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.B.; McClellan, M.L.; Reed, B.G. Sex differences, gender and addiction. J. Neurosci. Res. 2016, 95, 136–147. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Buck, L.A.; Bryant, K.G.; Barker, J.M. Sex Differences in Ethanol Reward Seeking Under Conflict in Mice. Alcohol. Clin. Exp. Res. 2019, 43, 1556–1566. [Google Scholar] [CrossRef]
- Torres, O.V.; Walker, E.M.; Beas, B.S.; O’Dell, L.E. Female rats display enhanced rewarding effects of ethanol that are hormone dependent. Alcohol. Clin. Exp. Res. 2013, 38, 108–115. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Crews, F.T. Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp. Neurol. 2008, 210, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Romanazzi, V.; Schilirò, T.; Carraro, E.; Gilli, G. Immune response to acetaldehyde-human serum albumin adduct among healthy subjects related to alcohol intake. Environ. Toxicol. Pharmacol. 2013, 36, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Petrasek, J.; Iracheta-Vellve, A.; Saha, B.; Satishchandran, A.; Kodys, K.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Szabo, G. Metabolic danger signals, uric acid and ATP, mediate inflammatory cross-talk between hepatocytes and immune cells in alcoholic liver disease. J. Leukoc. Boil. 2015, 98, 249–256. [Google Scholar] [CrossRef]
- De Santis, S.; Cosa-Linan, A.; Garcia-Hernandez, R.; Dmytrenko, L.; Vargova, L.; Vorisek, I.; Stopponi, S.; Bach, P.; Kirsch, P.; Kiefer, F.; et al. Chronic alcohol consumption alters extracellular space geometry and transmitter diffusion in the brain. Sci. Adv. 2020, 6, eaba0154. [Google Scholar] [CrossRef] [PubMed]
- Nissen, J.C. Microglial Function across the Spectrum of Age and Gender. Int. J. Mol. Sci. 2017, 18, 561. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Della Torre, S.; Maggi, A. Sexual differentiation of microglia. Front. Neuroendocr. 2019, 52, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Pavarin, R.M.; Sanchini, S.; Marani, S.; Turino, E.; Tadonio, L.; Caputo, F. Mortality risk among individuals treated for alcohol use disorders: Results of a longitudinal study from 1978 to 2016 in Northern Italy. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 1995–2005. [Google Scholar]
- Day, E.; Rudd, J.H.F. Alcohol use disorders and the heart. Addiction 2019, 114, 1670–1678. [Google Scholar] [CrossRef] [Green Version]
- Hitzemann, R.; Phillips, T.J.; Lockwood, D.R.; Darakjian, P.; Searles, R.P. Phenotypic and gene expression features associated with variation in chronic ethanol consumption in heterogeneous stock collaborative cross mice. Genomics 2020, 112, 4516–4525. [Google Scholar] [CrossRef]
- Dennis, M.K.; Delevoye, C.; Acosta-Ruiz, A.; Hurbain, I.; Romao, M.; Hesketh, G.G.; Goff, P.S.; Sviderskaya, E.V.; Bennett, D.C.; Luzio, J.P.; et al. BLOC-1 and BLOC-3 regulate VAMP7 cycling to and from melanosomes via distinct tubular transport carriers. J. Cell Boil. 2016, 214, 293–308. [Google Scholar] [CrossRef] [Green Version]
- Chanaday, N.L.; Cousin, M.A.; Milosevic, I.; Watanabe, S.; Morgan, J.R. The Synaptic Vesicle Cycle Revisited: New Insights into the Modes and Mechanisms. J. Neurosci. 2019, 39, 8209–8216. [Google Scholar] [CrossRef] [Green Version]
- Mooney, S.M.; Miller, M.W. Time-specific effects of ethanol exposure on cranial nerve nuclei: Gastrulation and neuronogenesis. Exp. Neurol. 2007, 205, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Gill, J. THE EFFECTS OF MODERATE ALCOHOL CONSUMPTION ON FEMALE HORMONE LEVELS AND REPRODUCTIVE FUNCTION. Alcohol Alcohol. 2000, 35, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Guerri, C.; Pascual, M. Chapter 24: Effects of Alcohol on Embryo/Fetal Development. In Reproductive and Developmental Toxicology, 2nd ed.; Gupta, R.C., Ed.; Academic Press: Cambridge, MA, USA, 2017; ISBN 9780123820327. [Google Scholar]
- Schaffer, A.E.; Pinkard, O.; Coller, J.M. tRNA Metabolism and Neurodevelopmental Disorders. Annu. Rev. Genom. Hum. Genet. 2019, 20, 359–387. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO Accession | Platform | Number of Samples | Sample Tissue | Citation |
|---|---|---|---|---|
| GSE44456 1 | GPL6244 Affymetrix Human Gene 1.0 ST Array | 39 | Hippocampus | McClintick, J. et al. [33] |
| GSE49376 2 | GPL10904 Illumina HumanHT-12 V4.0 expression beadchip | 48 | Dorsolateral prefrontal cortex | Xu, H. et al. [34] |
| GSE52553 3 | GPL570 Affymetrix Human Genome U133 Plus 2.0 Array | 42 | Immortalized lymphoblasts from blood samples | McClintick, J. et al. [37] |
| GSE59206 4 | GPL10558 Illumina HumanHT-12 V4.0 expression beadchip | 22 | Whole blood | Beech, R. et al. [39] |
| GO Terms | KEGG Pathways | |||
|---|---|---|---|---|
| Studies | Positive LOR | Negative LOR | Positive LOR | Negative LOR |
| GSE44456 1 | 1208 | 703 | 39 | 25 |
| GSE49376 2 | 449 | 802 | 16 | 25 |
| GSE52553 3 | 7 | 66 | 0 | 2 |
| GSE59206 4 | 113 | 14 | 5 | 0 |
| Ontology/Database | Positive LOR | Negative LOR |
|---|---|---|
| Biological Processes | 134 | 151 |
| Cellular Components | 73 | 23 |
| Molecular Functions | 55 | 24 |
| KEGG pathways | 5 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casanova Ferrer, F.; Pascual, M.; Hidalgo, M.R.; Malmierca-Merlo, P.; Guerri, C.; García-García, F. Unveiling Sex-Based Differences in the Effects of Alcohol Abuse: A Comprehensive Functional Meta-Analysis of Transcriptomic Studies. Genes 2020, 11, 1106. https://doi.org/10.3390/genes11091106
Casanova Ferrer F, Pascual M, Hidalgo MR, Malmierca-Merlo P, Guerri C, García-García F. Unveiling Sex-Based Differences in the Effects of Alcohol Abuse: A Comprehensive Functional Meta-Analysis of Transcriptomic Studies. Genes. 2020; 11(9):1106. https://doi.org/10.3390/genes11091106
Chicago/Turabian StyleCasanova Ferrer, Franc, María Pascual, Marta R. Hidalgo, Pablo Malmierca-Merlo, Consuelo Guerri, and Francisco García-García. 2020. "Unveiling Sex-Based Differences in the Effects of Alcohol Abuse: A Comprehensive Functional Meta-Analysis of Transcriptomic Studies" Genes 11, no. 9: 1106. https://doi.org/10.3390/genes11091106
APA StyleCasanova Ferrer, F., Pascual, M., Hidalgo, M. R., Malmierca-Merlo, P., Guerri, C., & García-García, F. (2020). Unveiling Sex-Based Differences in the Effects of Alcohol Abuse: A Comprehensive Functional Meta-Analysis of Transcriptomic Studies. Genes, 11(9), 1106. https://doi.org/10.3390/genes11091106