Integrated Analysis of Gene Expression, SNP, InDel, and CNV Identifies Candidate Avirulence Genes in Australian Isolates of the Wheat Leaf Rust Pathogen Puccinia triticina


Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Sequencing
2.2. RNA Sequencing
2.3. RNA-Seq Analysis for the Selection of Expressed Effectors
2.4. Quality Assessing, Trimming, and Mapping for Whole-Genome Sequencing
2.5. SNP and InDel Calling and Comparative Genomic Analysis
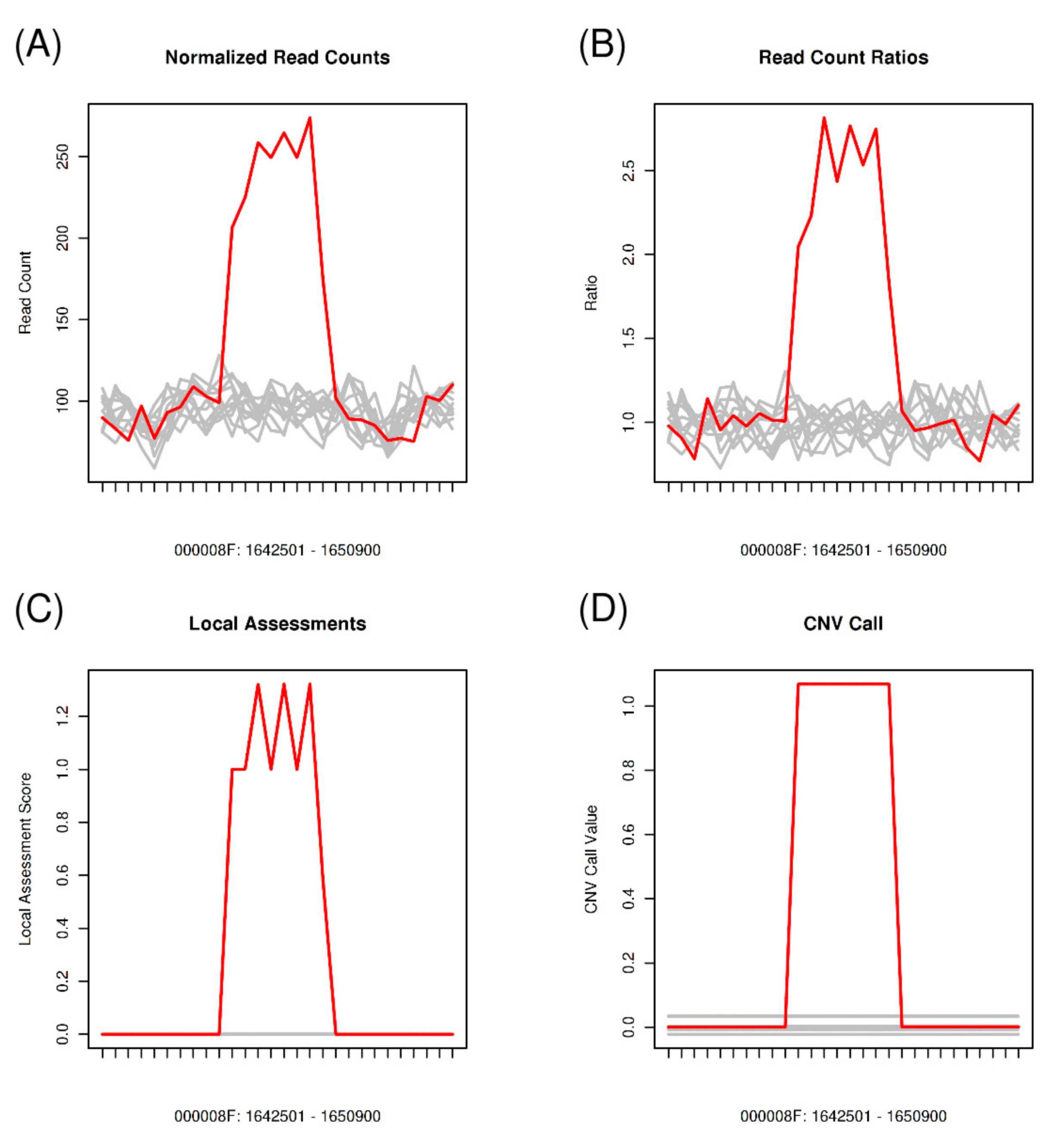
2.6. Copy Number Variation Analysis
3. Results
3.1. Secretome Prediction by EffectorP and Validation by RNA Sequencing
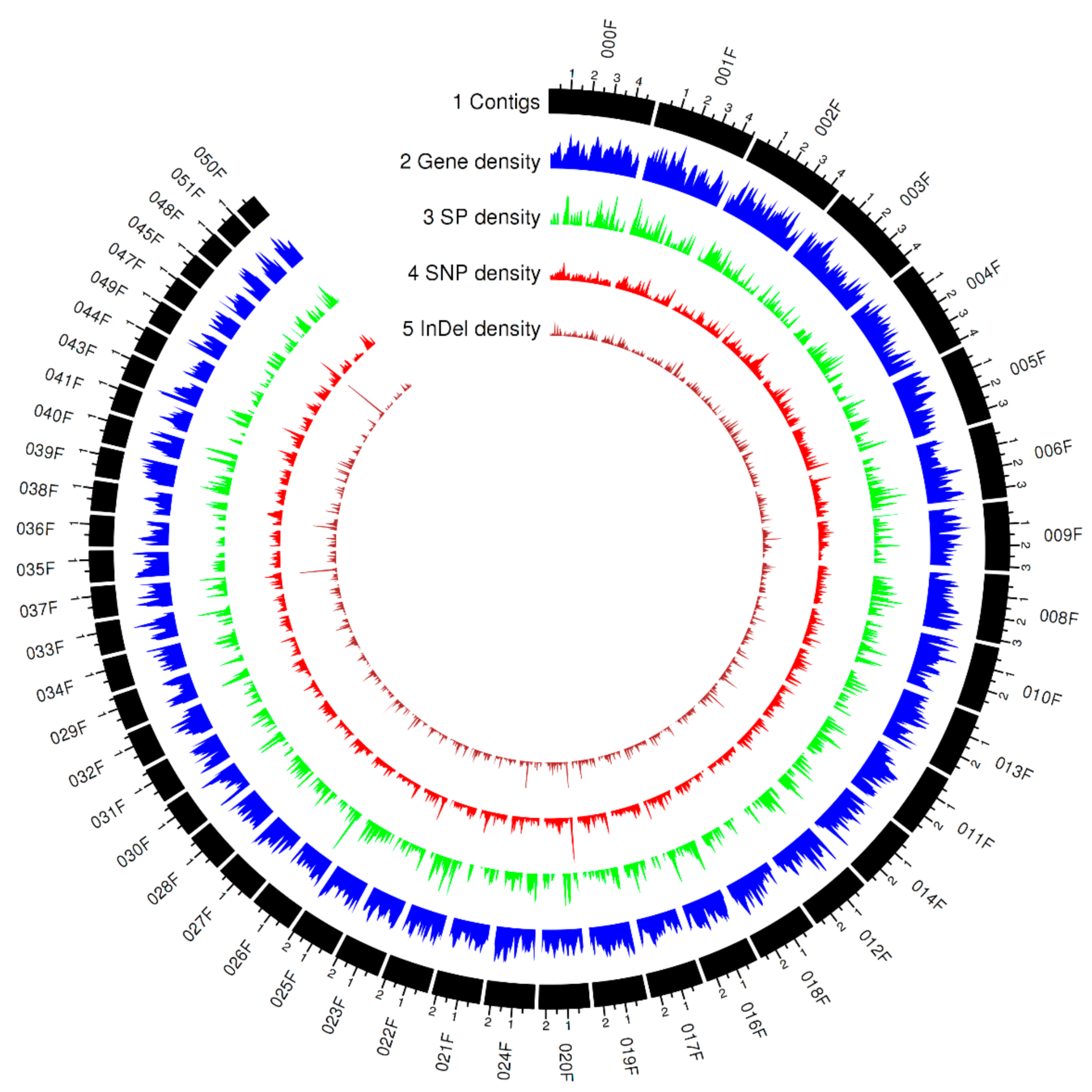
3.2. Mapping Whole-Genome Sequencing Data
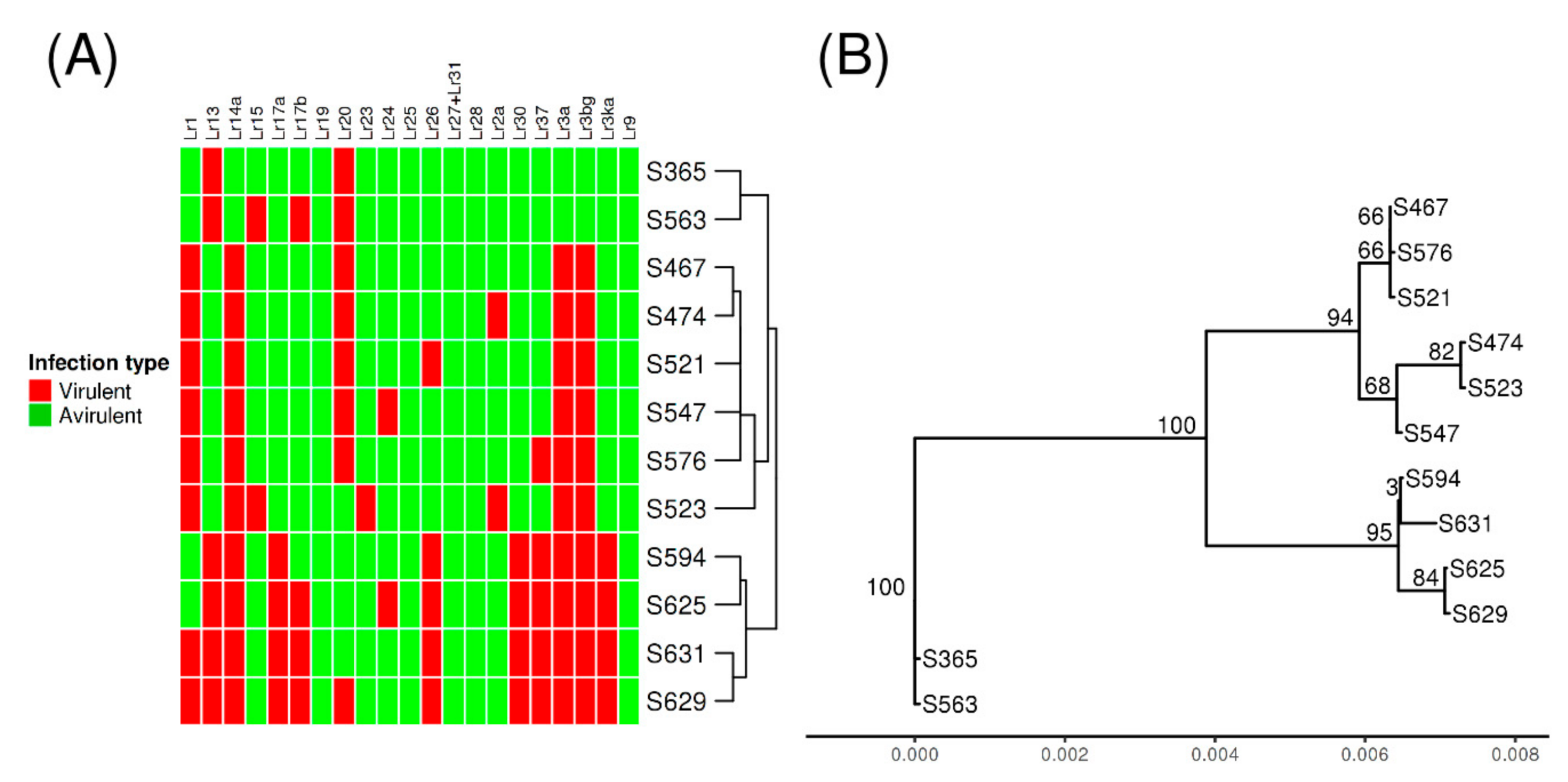
3.3. Genome-Wide Polymorphism and Phylogenetic Analysis
3.4. Functional Impact of Small Genomic Variants
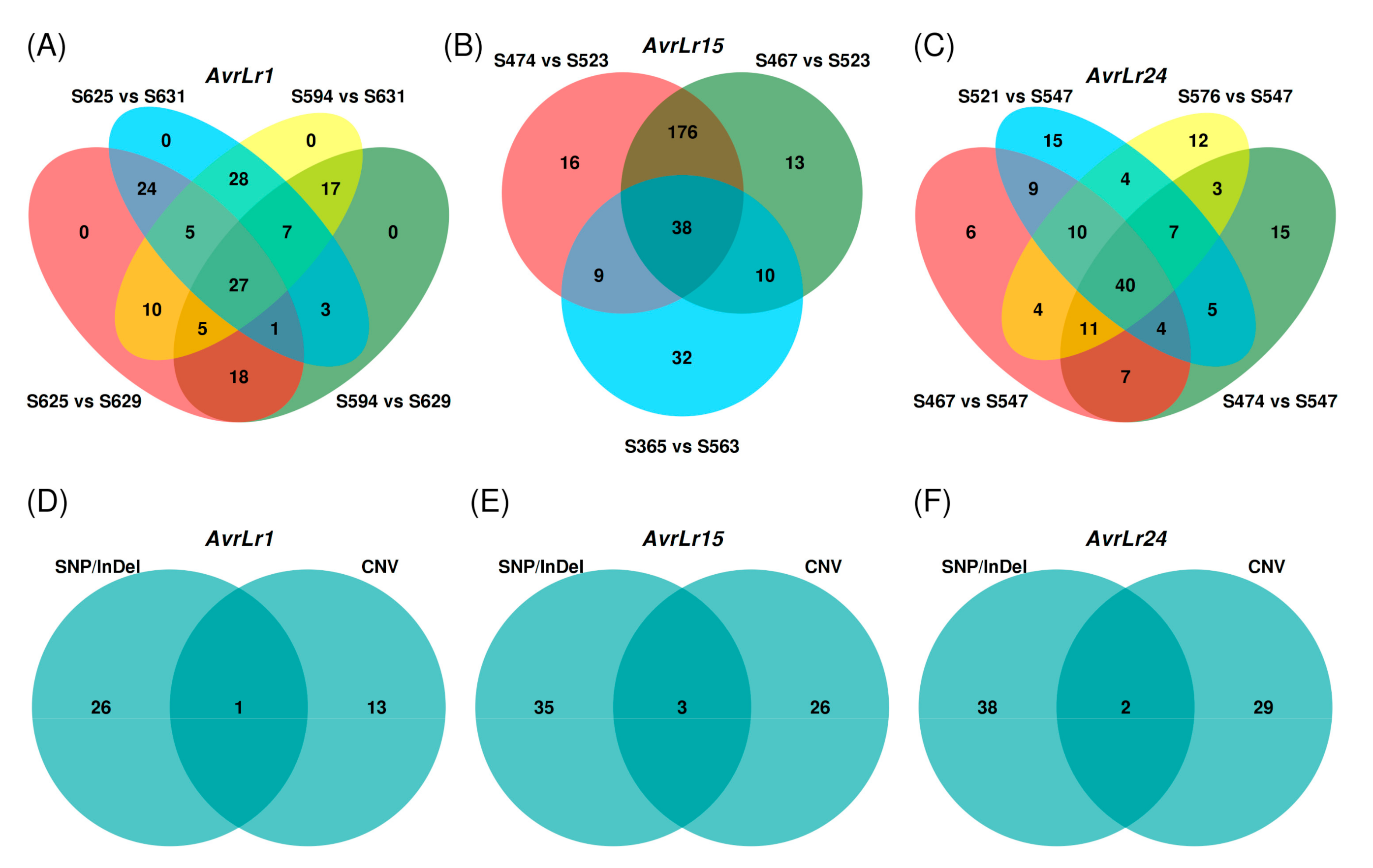
3.5. Small Genomic Variations Correlated with Avirulence/Virulence Phenotype
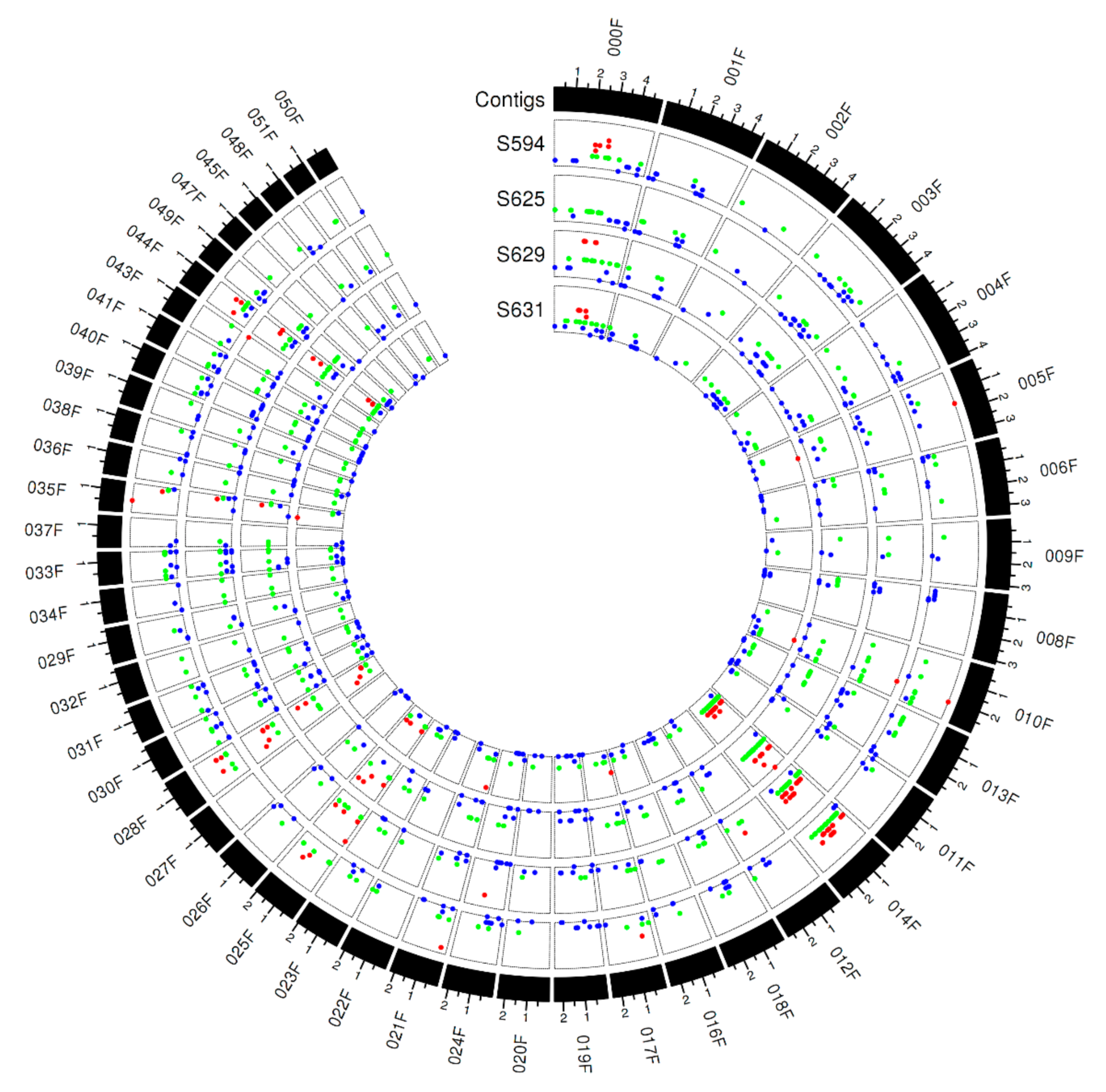
3.6. Copy Number Variations across the Pt Isolates
3.7. Copy Number Variations Correlated with Avirulence/Virulence Phenotype
3.8. Final Candidate Avirulence Genes and Their Biological Annotations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Savary, S.; Willocquet, L.; Pethybridge, S.J.; Esker, P.; McRoberts, N.; Nelson, A. The global burden of pathogens and pests on major food crops. Nat. Ecol. Evol. 2019, 3, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Park, R.F. Breeding cereals for rust resistance in Australia. Plant Pathol. 2008, 57, 591–602. [Google Scholar] [CrossRef]
- Bolton, M.D.; Kolmer, J.A.; Garvin, D.F. Wheat leaf rust caused by Puccinia triticina. Mol. Plant Pathol. 2008, 9, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Kolmer, J.A. Leaf Rust of Wheat: Pathogen Biology, Variation and Host Resistance. Forests 2013, 4, 70–84. [Google Scholar] [CrossRef] [Green Version]
- Kolmer, J.A.; Hughes, M.E. Physiologic Specialization of Puccinia triticina on Wheat in the United States in 2014. Plant Dis. 2016, 100, 1768–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flor, H.H. Current Status of the Gene-For-Gene Concept. Annu. Rev. Phytopathol. 1971, 9, 275–296. [Google Scholar] [CrossRef]
- Jones, J.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Dodds, P.N.; Lawrence, G.J.; Catanzariti, A.-M.; Teh, T.; Wang, C.-I.A.; Ayliffe, M.A.; Kobe, B.; Ellis, J.G. Direct protein interaction underlies gene-for-gene specificity and coevolution of the flax resistance genes and flax rust avirulence genes. Proc. Natl. Acad. Sci. USA 2006, 103, 8888–8893. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-S.; Desveaux, D.; Singer, A.U.; Patel, P.; Sondek, J.; Dangl, J.L. The Pseudomonas syringae effector AvrRpt2 cleaves its C-terminally acylated target, RIN4, from Arabidopsis membranes to block RPM1 activation. Proc. Natl. Acad. Sci. USA 2005, 102, 6496–6501. [Google Scholar] [CrossRef] [Green Version]
- Aktar-Uz-Zaman, M.; Tuhina-Khatun, M.; Hanafi, M.M.; Sahebi, M. Genetic analysis of rust resistance genes in global wheat cultivars: An overview. Biotechnol. Biotechnol. Equip. 2017, 31, 431–445. [Google Scholar] [CrossRef] [Green Version]
- Prasad, P.; Savadi, S.; Bhardwaj, S.C.; Gangwar, O.P.; Kumar, S. Rust pathogen effectors: Perspectives in resistance breeding. Planta 2019, 250, 1–22. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, R.A.; Wellings, C.R.; Park, R.F. Wheat Rusts: An Atlas of Resistance Genes; CSIRO: Melbourne, Australia, 1995. [Google Scholar]
- Huerta-Espino, J.; Singh, R.P.; German, S.; McCallum, B.D.; Park, R.F.; Chen, W.Q.; Bhardwaj, S.C.; Goyeau, H. Global status of wheat leaf rust caused by Puccinia triticina. Euphytica 2011, 179, 143–160. [Google Scholar] [CrossRef]
- Zwer, P.; Park, R.F.; McIntosh, R. Wheat stem rust in Australia dash 1969–1985. Aust. J. Agric. Res. 1992, 43, 399–431. [Google Scholar] [CrossRef]
- Park, R.F. Rust Fungi. In Encyclopedia of Microbiology, 2nd ed.; Academic Press: San Diego, CA, USA, 2000; pp. 195–211. [Google Scholar]
- Park, R.F.; Burdon, J.J.; McIntosh, R.A. Studies on the origin, spread, and evolution of an important group of Puccinia recondita f. sp.tritici pathotypes in Australasia. Eur. J. Plant Pathol. 1995, 101, 613–622. [Google Scholar] [CrossRef]
- Park, R.F.; Burdon, J.; Jahoor, A. Evidence for somatic hybridization in nature in Puccinia recondita f. sp. tritici, the leaf rust pathogen of wheat. Mycol. Res. 1999, 103, 715–723. [Google Scholar] [CrossRef]
- Lorrain, C.; Dos Santos, K.C.G.; Germain, H.; Hecker, A.; Duplessis, S. Advances in understanding obligate biotrophy in rust fungi. New Phytol. 2019, 222, 1190–1206. [Google Scholar] [CrossRef] [Green Version]
- Cantu, D.; Segovia, V.; MacLean, D.; Bayles, R.; Chen, X.; Kamoun, S.; Dubcovsky, J.; Saunders, D.G.O.; Uauy, C. Genome analyses of the wheat yellow (stripe) rust pathogen Puccinia striiformis f. sp. tritici reveal polymorphic and haustorial expressed secreted proteins as candidate effectors. BMC Genom. 2013, 14, 270. [Google Scholar] [CrossRef] [Green Version]
- Kiran, K.; Rawal, H.C.; Dubey, H.; Jaswal, R.; Bhardwaj, S.C.; Prasad, P.; Pal, D.; Devanna, B.N.; Sharma, T.R. Dissection of genomic features and variations of three pathotypes of Puccinia striiformis through whole genome sequencing. Sci. Rep. 2017, 7, 42419. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xia, C.; Wang, M.; Yin, C.; Chen, X. Whole-genome sequencing of Puccinia striiformis f. sp. tritici mutant isolates identifies avirulence gene candidates. BMC Genom. 2020, 21, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, N.M.; Garnica, D.P.; Karaoglu, H.; Sperschneider, J.; Nemri, A.; Xu, B.; Mago, R.; Cuomo, C.A.; Rathjen, J.P.; Park, R.F.; et al. Comparative genomics of Australian isolates of the wheat stem rust pathogen Puccinia graminis f. sp. tritici reveals extensive polymorphism in candidate effector genes. Front. Plant Sci. 2015, 5, 759. [Google Scholar] [CrossRef]
- Wu, J.Q.; Sakthikumar, S.; Dong, C.; Zhang, P.; Cuomo, C.A.; Park, R.F. Comparative Genomics Integrated with Association Analysis Identifies Candidate Effector Genes Corresponding to Lr20 in Phenotype-Paired Puccinia triticina Isolates from Australia. Front. Plant Sci. 2017, 8, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, M.; Neugebauer, K.A.; Joly, D.; Migeon, P.; Cuomo, C.A.; Wang, S.; Akhunov, E.; Bakkeren, G.; Kolmer, J.A.; Fellers, J.P. Using transcription of six Puccinia triticina races to identify the effective secretome during infection of wheat. Front. Plant Sci. 2014, 4, 520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Upadhyaya, N.M.; Ortiz, D.; Sperschneider, J.; Li, F.; Bouton, C.R.; Breen, S.; Dong, C.M.; Xu, B.; Zhang, X.; et al. Loss of AvrSr50 by somatic exchange in stem rust leads to virulence for Sr50 resistance in wheat. Science 2017, 358, 1607–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salcedo, A.; Rutter, W.; Wang, S.; Akhunov, A.; Bolus, S.; Chao, S.; Anderson, N.; De Soto, M.F.; Rouse, M.N.; Szabo, L.J.; et al. Variation in the AvrSr35 gene determines Sr35 resistance against wheat stem rust race Ug99. Science 2017, 358, 1604–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.Q.; Dong, C.; Song, L.; Park, R.F. Long-Read–Based de novo Genome Assembly and Comparative Genomics of the Wheat Leaf Rust Pathogen Puccinia triticina Identifies Candidates for Three Avirulence Genes. Front. Genet. 2020, 11, 521. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wu, J.; Zhang, P.; Dong, C.; Upadhyaya, N.M.; Zhou, Q.; Dodds, P.; Park, R.F. De Novo Genome Assembly and Comparative Genomics of the Barley Leaf Rust Pathogen Puccinia hordei Identifies Candidates for Three Avirulence Genes. G3 Genes Genomes Genet. 2019, 9, 3263–3271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steenwyk, J.L.; Rokas, A. Copy Number Variation in Fungi and Its Implications for Wine Yeast Genetic Diversity and Adaptation. Front. Microbiol. 2018, 9, 288. [Google Scholar] [CrossRef] [Green Version]
- Steenwyk, J.L.; Rokas, A. Extensive Copy Number Variation in Fermentation-Related Genes among Saccharomyces cerevisiae Wine Strains. G3 Genes Genomes Genet. 2017, 7, 1475–1485. [Google Scholar] [CrossRef] [Green Version]
- Strope, P.K.; Skelly, D.A.; Kozmin, S.G.; Mahadevan, G.; Stone, E.A.; Magwene, P.M.; Dietrich, F.S.; McCusker, J.H. The 100-genomes strains, an S. cerevisiae resource that illuminates its natural phenotypic and genotypic variation and emergence as an opportunistic pathogen. Genome Res. 2015, 25, 762–774. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, F.E.; Croll, D. Distinct Trajectories of Massive Recent Gene Gains and Losses in Populations of a Microbial Eukaryotic Pathogen. Mol. Biol. Evol. 2017, 34, 2808–2822. [Google Scholar] [CrossRef] [Green Version]
- Steenwyk, J.L.; Soghigian, J.S.; Perfect, J.R.; Gibbons, J.G. Copy number variation contributes to cryptic genetic variation in outbreak lineages of Cryptococcus gattii from the North American Pacific Northwest. BMC Genom. 2016, 17, 700. [Google Scholar] [CrossRef] [Green Version]
- Schwessinger, B.; Rathjen, J.P. Extraction of High Molecular Weight DNA from Fungal Rust Spores for Long Read Sequencing. Adv. Struct. Saf. Stud. 2017, 1659, 49–57. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperschneider, J.; Dodds, P.N.; Gardiner, D.M.; Singh, K.B.; Taylor, J.M. Improved prediction of fungal effector proteins from secretomes with EffectorP 2.0. Mol. Plant Pathol. 2018, 19, 2094–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechner, M.; Findeiss, S.; Mueller, L.; Marz, M.; Stadler, P.F.; Prohaska, S. Proteinortho: Detection of (Co-)orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperschneider, J.; Catanzariti, A.-M.; DeBoer, K.; Petre, B.; Gardiner, D.M.; Singh, K.B.; Dodds, P.N.; Taylor, J.M. LOCALIZER: Subcellular localization prediction of both plant and effector proteins in the plant cell. Sci. Rep. 2017, 7, 44598. [Google Scholar] [CrossRef] [Green Version]
- Sperschneider, J.; Dodds, P.N.; Singh, K.B.; Taylor, J.M. ApoplastP: Prediction of effectors and plant proteins in the apoplast using machine learning. New Phytol. 2017, 217, 1764–1778. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 21 June 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Van Der Auwera, G.A.; Carneiro, M.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 1–33. [Google Scholar] [CrossRef]
- Poplin, R.; Ruano-Rubio, V.; De Pristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2017, 201178. [Google Scholar] [CrossRef] [Green Version]
- Garrison, E. Vcflib, a Simple C++ Library for Parsing and Manipulating VCF Files. 2016. Available online: https://github.com/vcflib/vcflib (accessed on 21 June 2020).
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.-H.; Guo, H.; Wang, X.; Kim, C.; Paterson, A.H. SNPhylo: A pipeline to construct a phylogenetic tree from huge SNP data. BMC Genom. 2014, 15, 162. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Lam, T.T.-Y.; Zhu, H.; Guan, Y. Two Methods for Mapping and Visualizing Associated Data on Phylogeny Using Ggtree. Mol. Biol. Evol. 2018, 35, 3041–3043. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [Green Version]
- Obenchain, V.; Lawrence, M.; Carey, V.; Gogarten, S.M.; Shannon, P.; Morgan, M.T. VariantAnnotation: A Bioconductor package for exploration and annotation of genetic variants. Bioinformatics 2014, 30, 2076–2078. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.; Gentleman, R.; Carey, V. rtracklayer: An R package for interfacing with genome browsers. Bioinformatics 2009, 25, 1841–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]
- Klambauer, G.; Schwarzbauer, K.; Mayr, A.; Clevert, D.-A.; Mitterecker, A.; Bodenhofer, U.; Hochreiter, S. cn.MOPS: Mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res. 2012, 40, e69. [Google Scholar] [CrossRef] [PubMed]
- Cuomo, C.A.; Bakkeren, G.; Khalil, H.B.; Panwar, V.; Joly, D.; Linning, R.; Sakthikumar, S.; Song, X.; Adiconis, X.; Fan, L.; et al. Comparative Analysis Highlights Variable Genome Content of Wheat Rusts and Divergence of the Mating Loci. G3 Genes Genomes Genet. 2017, 7, 361–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, G.; Wang, J.; Choi, J.; Jung, W.H.; Liu, I.; Litvintseva, A.P.; Bicanic, T.; Aurora, R.; Mitchell, T.G.; Perfect, J.R.; et al. Variation in chromosome copy number influences the virulence of Cryptococcus neoformans and occurs in isolates from AIDS patients. BMC Genom. 2011, 12, 526. [Google Scholar] [CrossRef] [Green Version]
- Farrer, R.A.; Henk, D.A.; Garner, T.W.J.; Balloux, F.; Woodhams, D.C.; Fisher, M.C. Chromosomal Copy Number Variation, Selection and Uneven Rates of Recombination Reveal Cryptic Genome Diversity Linked to Pathogenicity. PLoS Genet. 2013, 9, e1003703. [Google Scholar] [CrossRef] [Green Version]
- Qutob, D.; Tedman-Jones, J.; Dong, S.; Kuflu, K.; Pham, H.; Wang, Y.; Dou, D.; Kale, S.D.; Arredondo, F.D.; Tyler, B.M.; et al. Copy number variation and transcriptional polymorphisms of Phytophthora sojae RXLR effector genes Avr1a and Avr3a. PLoS ONE 2009, 4, e5066. [Google Scholar] [CrossRef]
- Wind, M.; Reines, D. Transcription elongation factor SII. BioEssays 2000, 22, 327–336. [Google Scholar] [CrossRef]
- Bourras, S.; Kunz, L.; Xue, M.; Praz, C.R.; Müller, M.C.; Kälin, C.; Schläfli, M.; Ackermann, P.; Flückiger, S.; Parlange, F.; et al. The AvrPm3-Pm3 effector-NLR interactions control both race-specific resistance and host-specificity of cereal mildews on wheat. Nat. Commun. 2019, 10, 2292. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate | Total Reads (Quality Trimmed) | Reads Mapped to Reference | Percentage Mapped Reads | Average Coverage Fold | Percentage of Mapped Bases in Reference |
|---|---|---|---|---|---|
| S365 | 81,605,446 | 75,995,860 | 93.1% | 72.7 | 98.2% |
| S563 | 72,359,456 | 66,985,626 | 92.6% | 62.5 | 98.1% |
| S467 | 71,491,329 | 66,285,859 | 92.7% | 62.0 | 99.4% |
| S474 | 66,268,039 | 57,035,893 | 86.1% | 53.6 | 99.3% |
| S521 | 75,331,554 | 62,273,511 | 82.7% | 58.2 | 99.3% |
| S523 | 75,802,433 | 56,814,472 | 75.0% | 54.5 | 99.3% |
| S547 | 72,921,641 | 63,758,642 | 87.4% | 59.9 | 99.3% |
| S576 | 69,188,369 | 63,626,559 | 92.0% | 59.1 | 99.3% |
| S594 | 89,362,930 | 80,982,321 | 90.6% | 75.5 | 97.8% |
| S625 | 69,718,296 | 64,161,304 | 92.0% | 60.5 | 97.7% |
| S629 | 96,147,346 | 87,921,703 | 91.4% | 81.7 | 97.9% |
| S631 | 78,547,125 | 69,753,222 | 88.8% | 65.5 | 97.9% |
| Isolate | Total Variants | SNP | InDel | Insertion | Deletion | Heterozygous SNP | Heterozygous InDel | Percentage of Heterozygosity |
|---|---|---|---|---|---|---|---|---|
| S365 | 686,935 | 606,306 | 80,629 | 45,961 | 34,668 | 478,487 | 39,772 | 75.4% |
| S563 | 683,433 | 603,530 | 79,903 | 45,531 | 34,372 | 478,208 | 39,448 | 75.7% |
| S467 | 529,092 | 454,513 | 74,579 | 44,710 | 29,869 | 450,156 | 38,851 | 92.4% |
| S474 | 525,308 | 451,414 | 73,894 | 44,211 | 29,683 | 446,808 | 38,494 | 92.4% |
| S521 | 529,110 | 454,715 | 74,395 | 44,474 | 29,921 | 450,188 | 38,884 | 92.4% |
| S523 | 581,330 | 505,061 | 76,269 | 45,163 | 31,106 | 500,500 | 41,167 | 93.2% |
| S547 | 529,291 | 454,915 | 74,376 | 44,490 | 29,886 | 450,085 | 38,905 | 92.4% |
| S576 | 527,896 | 453,762 | 74,134 | 44,379 | 29,755 | 449,108 | 38,612 | 92.4% |
| S594 | 558,307 | 485,758 | 72,549 | 42,137 | 30,412 | 373,086 | 31,204 | 72.4% |
| S625 | 555,027 | 483,220 | 71,807 | 41,651 | 30,156 | 372,931 | 31,133 | 72.8% |
| S629 | 560,570 | 487,576 | 72,994 | 42,382 | 30,612 | 374,965 | 31,388 | 72.5% |
| S631 | 555,702 | 483,678 | 72,024 | 41,857 | 30,167 | 372,691 | 30,791 | 72.6% |
| Isolate | Coding Variants | Percentage of Coding Variants | Genes Covered | Synonymous Variants | Nonsynonymous Variants | Frameshift Variants | Nonsense Variants |
|---|---|---|---|---|---|---|---|
| S365 | 104,269 | 15.2% | 17,989 | 33,534 | 60,875 | 8136 | 1724 |
| S563 | 103,663 | 15.2% | 17,938 | 33,392 | 60,528 | 8036 | 1707 |
| S467 | 80,511 | 15.2% | 15,681 | 25,843 | 45,740 | 7734 | 1194 |
| S474 | 79,896 | 15.2% | 15,597 | 25,748 | 45,298 | 7634 | 1216 |
| S521 | 80,514 | 15.2% | 15,682 | 25,939 | 45,630 | 7747 | 1198 |
| S523 | 88,416 | 15.2% | 16,834 | 28,584 | 50,655 | 7788 | 1389 |
| S547 | 80,496 | 15.2% | 15,686 | 25,865 | 45,737 | 7688 | 1206 |
| S576 | 80,238 | 15.2% | 15,655 | 25,745 | 45,635 | 7647 | 1211 |
| S594 | 84,957 | 15.2% | 15,567 | 27,235 | 48,775 | 7521 | 1426 |
| S625 | 85,022 | 15.3% | 15,575 | 27,359 | 48,694 | 7543 | 1426 |
| S629 | 85,340 | 15.2% | 15,628 | 27,281 | 49,085 | 7544 | 1430 |
| S631 | 84,818 | 15.3% | 15,560 | 27,149 | 48,727 | 7516 | 1426 |
| Isolate | CNV Count | CNV Median Size (bp) | CNV Total Size (bp) | Percentage of Bases of Reference | Overlapping-Gene CNVs | Overlapping-SP Gene CNVs | Affected Genes | Affected SP Genes |
|---|---|---|---|---|---|---|---|---|
| S365 | 2231 | 1800 | 10,609,561 | 7.5% | 1021 | 59 | 2039 | 69 |
| S563 | 2235 | 1800 | 10,507,349 | 7.5% | 1018 | 58 | 2014 | 67 |
| S467 | 307 | 2100 | 2,381,715 | 1.7% | 154 | 13 | 428 | 18 |
| S474 | 324 | 2100 | 2,342,069 | 1.7% | 155 | 12 | 432 | 17 |
| S521 | 318 | 2100 | 2,202,295 | 1.6% | 152 | 12 | 389 | 16 |
| S523 | 324 | 1800 | 1,997,415 | 1.4% | 149 | 10 | 385 | 16 |
| S547 | 328 | 2100 | 2,418,223 | 1.7% | 147 | 16 | 410 | 24 |
| S576 | 310 | 2100 | 2,247,015 | 1.6% | 152 | 13 | 407 | 18 |
| S594 | 1713 | 2100 | 9,297,278 | 6.6% | 819 | 52 | 1731 | 63 |
| S625 | 1692 | 2100 | 9,263,378 | 6.6% | 824 | 56 | 1736 | 68 |
| S629 | 1688 | 2100 | 9,207,278 | 6.6% | 804 | 54 | 1710 | 66 |
| S631 | 1704 | 2100 | 9,066,903 | 6.5% | 807 | 50 | 1692 | 61 |
| Avr Gene | SNP/InDel Candidates | CNV Candidates | Overlapped Candidates | Final Candidates |
|---|---|---|---|---|
| AvrLr1 | 27 | 14 | 1 | 40 |
| AvrLr15 | 38 | 29 | 3 | 64 |
| AvrLr24 | 40 | 31 | 2 | 69 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, L.; Wu, J.Q.; Dong, C.M.; Park, R.F. Integrated Analysis of Gene Expression, SNP, InDel, and CNV Identifies Candidate Avirulence Genes in Australian Isolates of the Wheat Leaf Rust Pathogen Puccinia triticina. Genes 2020, 11, 1107. https://doi.org/10.3390/genes11091107
Song L, Wu JQ, Dong CM, Park RF. Integrated Analysis of Gene Expression, SNP, InDel, and CNV Identifies Candidate Avirulence Genes in Australian Isolates of the Wheat Leaf Rust Pathogen Puccinia triticina. Genes. 2020; 11(9):1107. https://doi.org/10.3390/genes11091107
Chicago/Turabian StyleSong, Long, Jing Qin Wu, Chong Mei Dong, and Robert F. Park. 2020. "Integrated Analysis of Gene Expression, SNP, InDel, and CNV Identifies Candidate Avirulence Genes in Australian Isolates of the Wheat Leaf Rust Pathogen Puccinia triticina" Genes 11, no. 9: 1107. https://doi.org/10.3390/genes11091107
APA StyleSong, L., Wu, J. Q., Dong, C. M., & Park, R. F. (2020). Integrated Analysis of Gene Expression, SNP, InDel, and CNV Identifies Candidate Avirulence Genes in Australian Isolates of the Wheat Leaf Rust Pathogen Puccinia triticina. Genes, 11(9), 1107. https://doi.org/10.3390/genes11091107