Evolutionary Analysis of Infectious Bronchitis Virus Reveals Marked Genetic Diversity and Recombination Events

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Clinical Information, Samples Collection, and Processing
2.3. Viral RNA Extraction and Genetic Characterization
2.4. S1 Coding Region Sequencing, Sequence Analysis, and Phylogeny
2.5. Antigenicity Prediction and 3D Visualization of Mutations
2.6. Evolutionary Analysis
3. Results
3.1. Virus Detection and Molecular Epidemiology of Infectious Bronchitis Virus
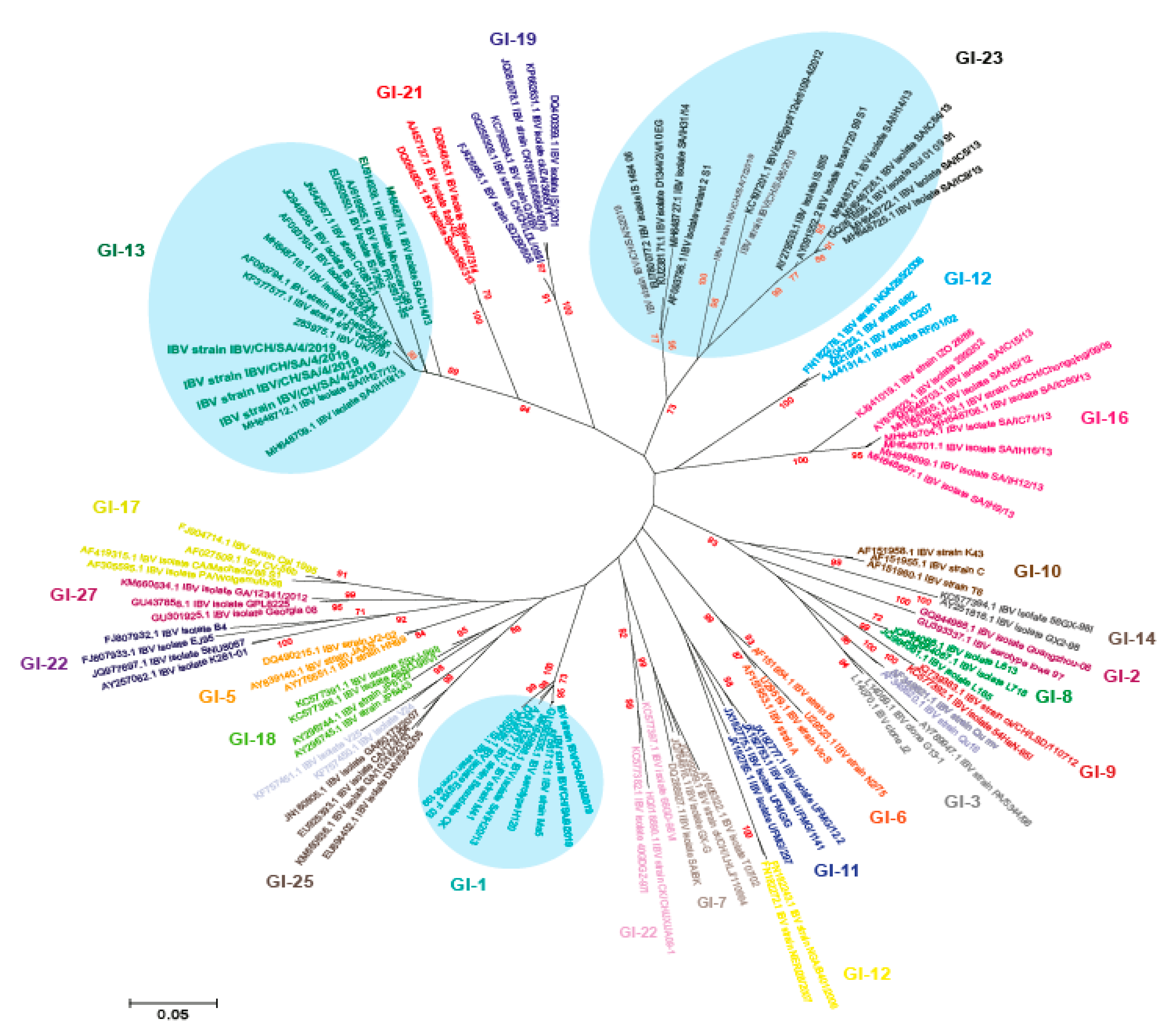
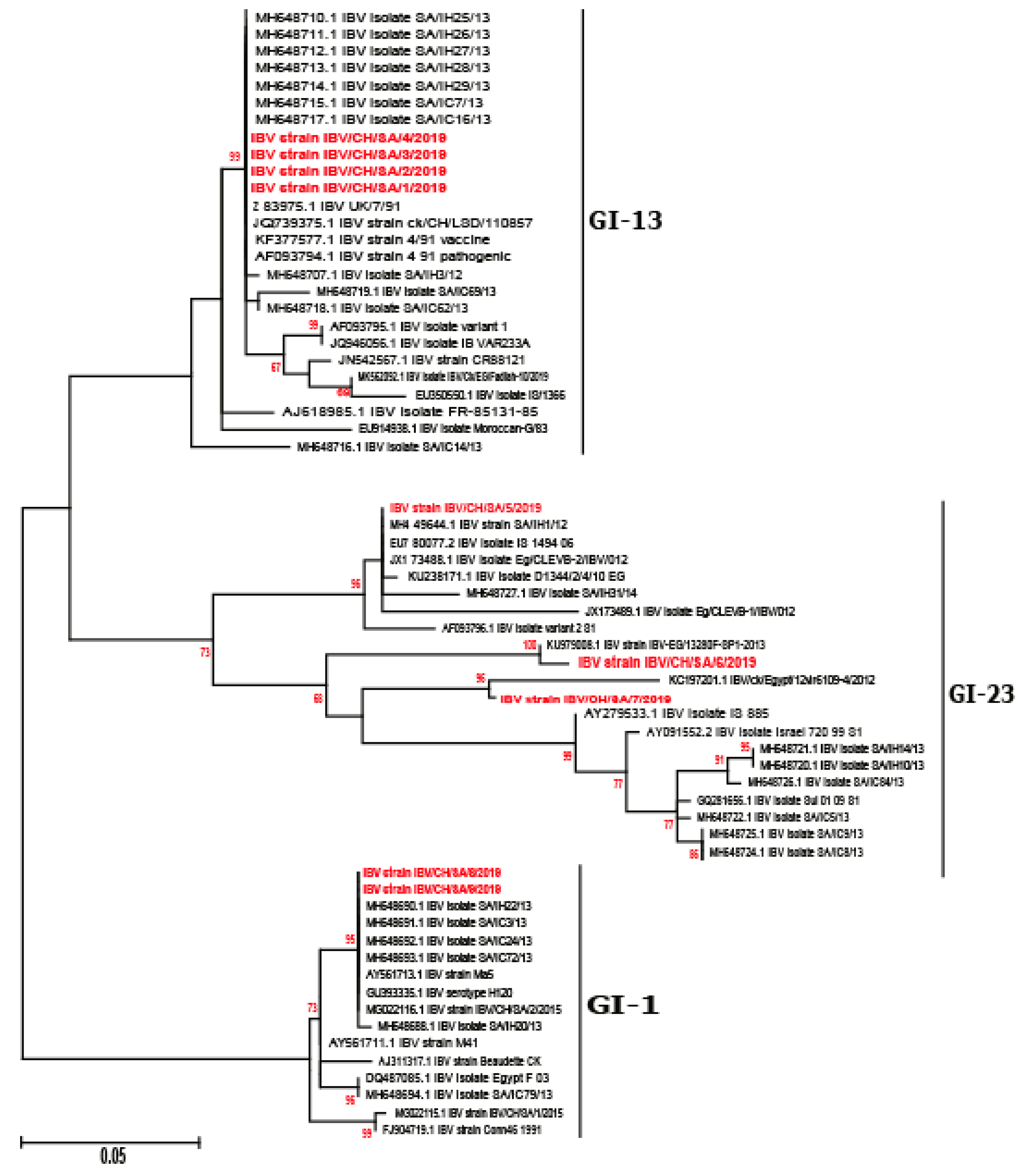
3.2. Phylogenetic Analysis
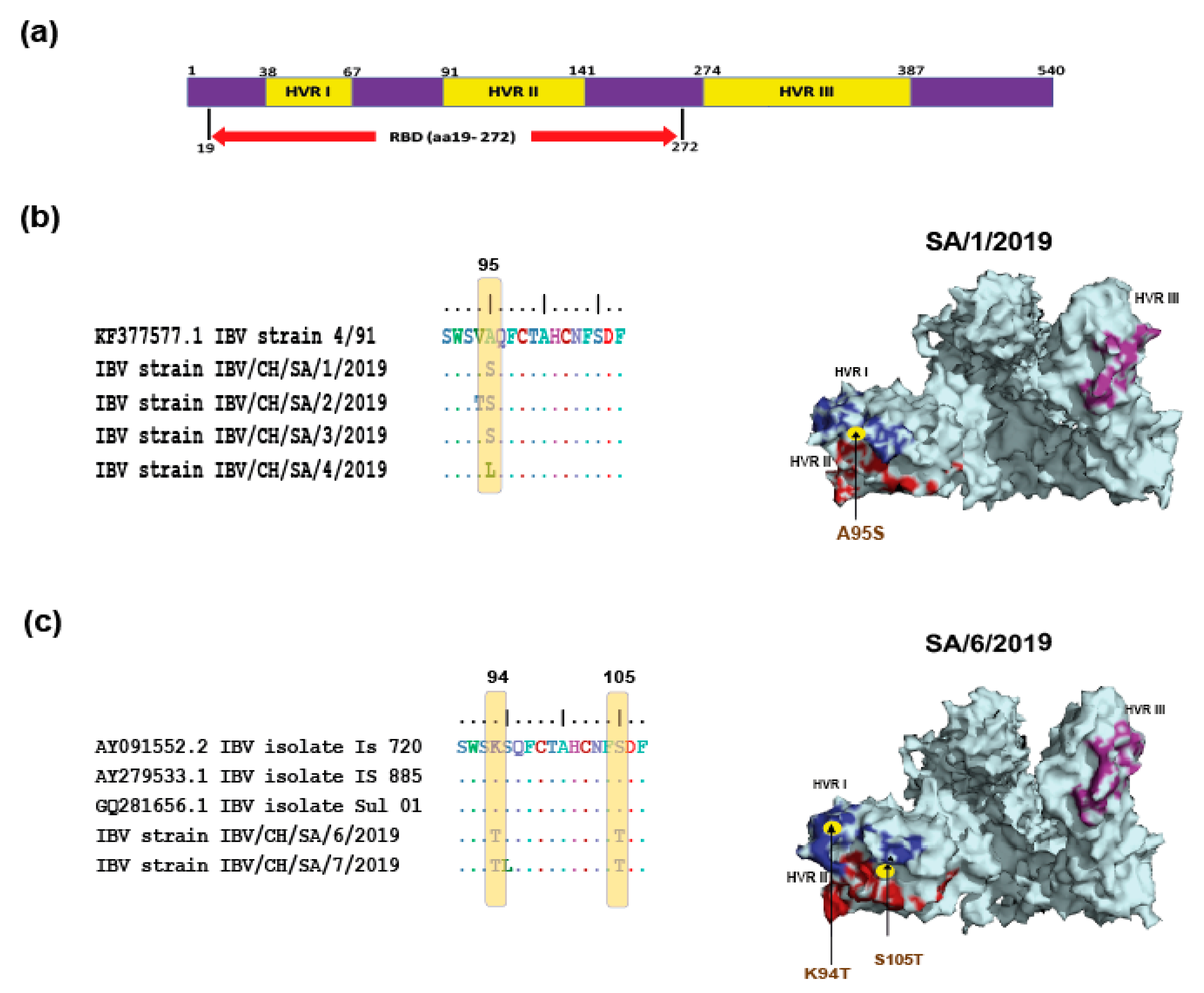
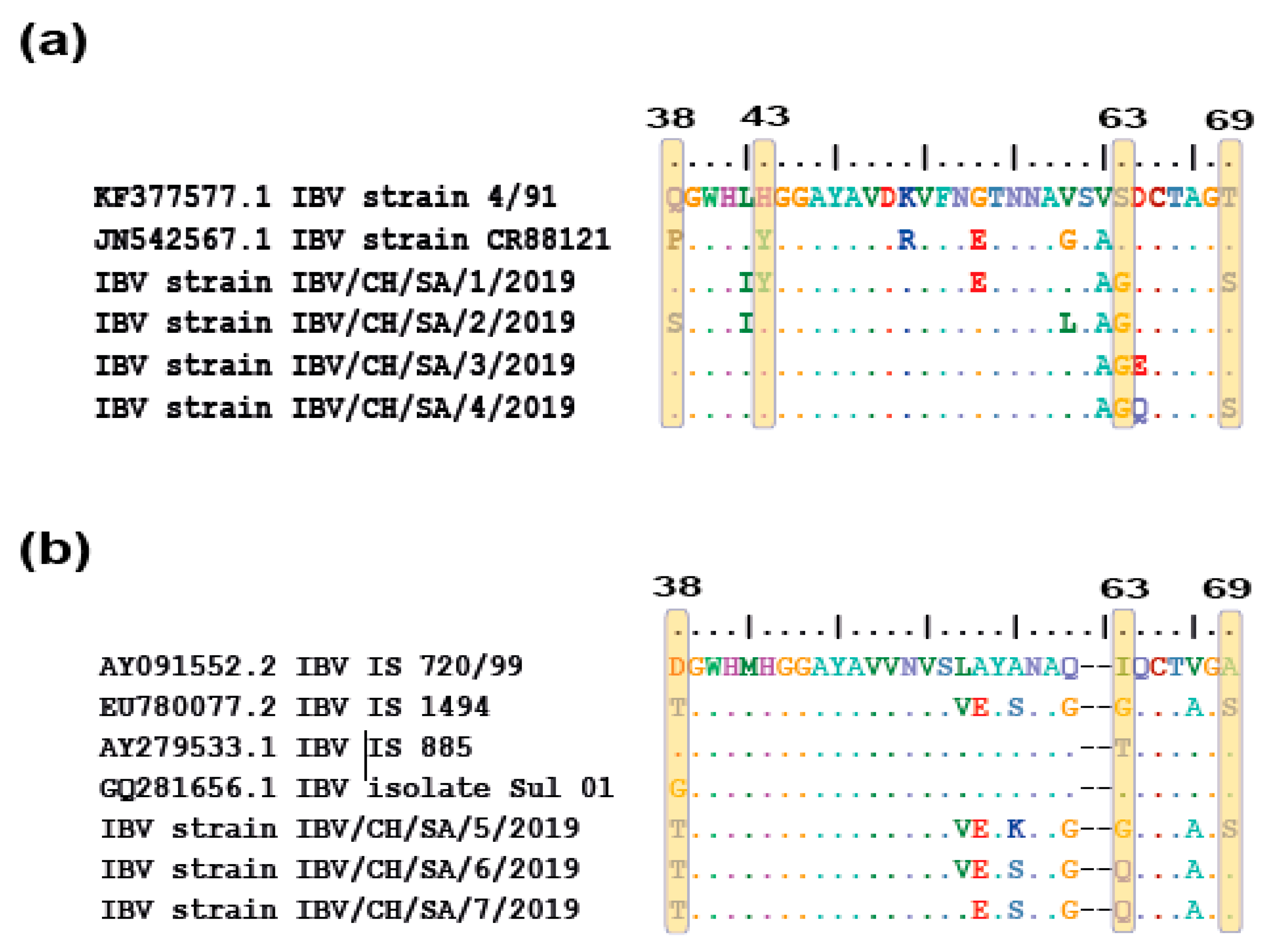
3.3. Comparative Amino Acid Analysis between Currently Circulating IBV Strains and Locally Used Vaccines
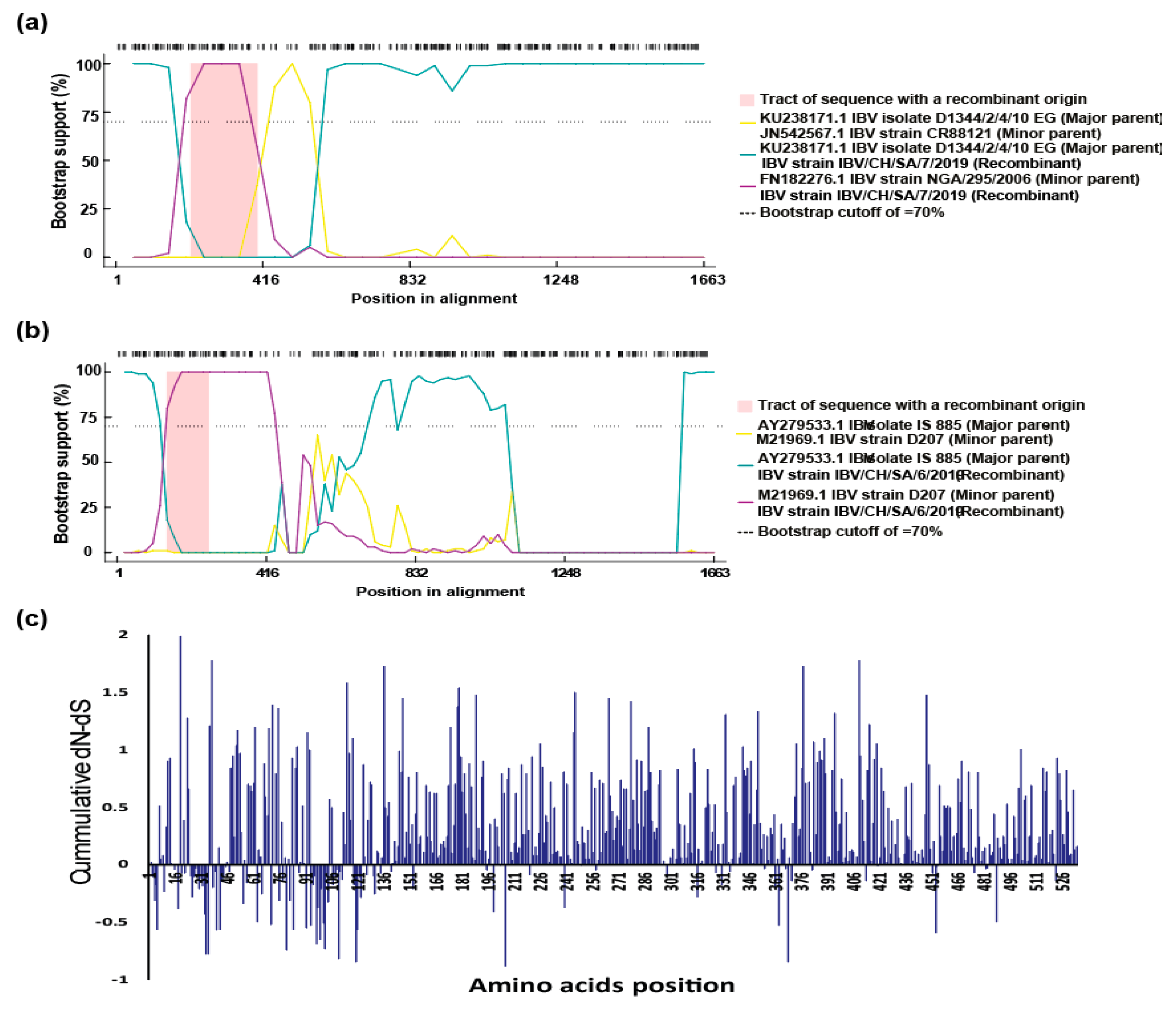
3.4. Recombination Analysis and Selective Pressure
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- de Groot, R. Family Coronaviridae. In Virus Taxonomy, 9th Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier Academic Press: San Diego, CA, USA, 2012; pp. 806–828. [Google Scholar]
- Cavanagh, D. Coronavirus avian infectious bronchitis virus. Vet. Res. 2007, 38, 281–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Promkuntod, N.; van Eijndhoven, R.E.; de Vrieze, G.; Gröne, A.; Verheije, M.H. Mapping of the receptor-binding domain and amino acids critical for attachment in the spike protein of avian coronavirus infectious bronchitis virus. Virology 2014, 448, 26–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavanagh, D.; Elus, M.M.; Cook, J.K. Relationship between sequence variation in the S1 spike protein of infectious bronchitis virus and the extent of cross-protection in vivo. Avian Pathol. 1997, 26, 63–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelb, J., Jr.; Weisman, Y.; Ladman, B.S.; Meir, R. S1 gene characteristics and efficacy of vaccination against infectious bronchitis virus field isolates from the United States and Israel (1996 to 2000). Avian Pathol. 2005, 34, 194–203. [Google Scholar] [CrossRef]
- Mockett, A.P.; Cavanagh, D.; Brown, T.D. Monoclonal antibodies to the S1 spike and membrane proteins of avian infectious bronchitis coronavirus strain Massachusetts M41. J. Gen. Virol. 1984, 65, 2281–2286. [Google Scholar] [CrossRef]
- Cavanagh, D.; Davis, P.J. Coronavirus IBV: Removal of spike glycopolypeptide S1 by urea abolishes infectivity and haemagglutination but not attachment to cells. J. Gen. Virol. 1986, 67, 1443–1448. [Google Scholar] [CrossRef]
- Cavanagh, D.; Davis, P.J.; Cook, J.K. Location of the amino acid differences in the S1 spike glycoprotein subunit of closely related serotypes of infectious bronchitis virus. Avian Pathol. 1992, 21, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.W.; Hilt, D.A.; Jackwood, M.W. Typing of field isolates of infectious bronchitis virus based on the sequence of the hypervariable region in the S1 gene. J. Vet. Diagn. Investig. 2003, 15, 344–348. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Junker, D.; Hock, L.; Ebiary, E.; Collisson, E.W. Evolutionary implications of genetic variations in the S1 gene of infectious bronchitis virus. Virus Res. 1994, 34, 327–338. [Google Scholar] [CrossRef]
- Cavanagh, D.; Gelb, J. Infectious bronchitis. In Diseases of Poultry, 12th ed.; Saif, Y.M., Fadly, A.M., Glisson, J.R., McDougald, L.R., Nolan, L.K., Swayne, D.E., Eds.; Blackwell Publishing Professional: Ames, IA, USA, 2008; p. 117135. [Google Scholar]
- Bande, F.; Arshad, S.S.; Omar, A.R.; Hair-Bejo, M.; Mahmuda, A.; Nair, V. Global distributions and strain diversity of avian infectious bronchitis virus: A review. Anim. Health Res. Rev. 2017, 18, 70–83. [Google Scholar] [CrossRef] [Green Version]
- Jackwood, M.W. Review of infectious bronchitis virus around the world. Avian Dis. 2012, 56, 634–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wit, J.J.; Nieuwenhuisen-van Wilgen, J.; Hoogkamer, A.; van de Sande, H.; Zuidam, G.J.; Fabri, T.H. Induction of cystic oviducts and protection against early challenge with infectious bronchitis virus serotype D388 (genotype QX) by maternally derived antibodies and by early vaccination. Avian Pathol. 2011, 40, 463–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, B. Vaccination against infectious bronchitis virus: A continuous challenge. Vet. Microbiol. 2017, 206, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Chen, H.W. Infectious bronchitis virus variants: Molecular analysis and pathogenicity investigation. Int. J. Mol. Sci. 2017, 18, 2030. [Google Scholar] [CrossRef] [Green Version]
- Valastro, V.; Holmes, E.C.; Britton, P.; Fusaro, A.; Jackwood, M.W.; Cattoli, G.; Isabella, M. S1 gene-based phylogeny of infectious bronchitis virus: An attempt to harmonize virus classification. Infect. Genet. Evol. 2016, 39, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jiang, L.; Zhao, W.; Liu, L.; Zhao, Y.; Shao, Y.; Li, H.; Han, Z.; Liu, S. Identification and molecular characterization of a novel serotype infectious bronchitis virus (GI-28) in China. Vet. Microbiol. 2017, 198, 108–115. [Google Scholar] [CrossRef]
- Jiang, L.; Han, Z.; Chen, Y.; Zhao, W.; Sun, J.; Zhao, Y.; Liu, S. Characterization of the complete genome, antigenicity, pathogenicity, tissue tropism, and shedding of a recombinant avian infectious bronchitis virus with a ck/CH/LJL/140901-like backbone and an S2 fragment from a 4/91-like virus. Virus Res. 2018, 244, 99–109. [Google Scholar] [CrossRef]
- Office International des Epizooties. Manual Diagnostic Tests Vaccines Terrestrial Animals (Mammals, Birds Bees); Office International des Epizooties: Paris, France, 2018. [Google Scholar]
- Meir, R.; Maharat, O.; Farnushi, Y.; Simanov, L. Development of a real-time TaqMant RT-PCR assay for the detection of infectious bronchitis virus in chickens, and comparison of RT-PCR and virus isolation. J. Virol. Methods 2010, 163, 190–194. [Google Scholar] [CrossRef]
- Boursnell, M.E.G.; Brown, T.D.K.; Foulds, I.J.; Green, P.F.; Tomley, F.M.; Binns, M.M. Completion of the sequence of the genome of the Coronavirus avian infectious bronchitis virus. J. Gen. Virol. 1987, 68, 57–77. [Google Scholar] [CrossRef]
- Dolz, R.; Pujols, J.; Ordonez, G.; Porta, R.; Majo, N. Antigenic and molecular characterization of isolates of the Italy 02 infectious bronchitis virus genotype. Avian Pathol. 2006, 35, 7785. [Google Scholar] [CrossRef]
- Worthington, K.J.; Currie, R.J.; Jones, R.C. A reverse transcriptase-polymerase chain reaction survey of infectious bronchitis virus genotypes in Western Europe from 2002 to 2006. Avian Pathol. 2008, 37, 247257. [Google Scholar] [CrossRef] [Green Version]
- Lisowska, A.; Sajewicz-Krukowska, J.; Fusaro, A.; Pikula, A.; Domanska-Blicharz, K. First characterization of a Middle-EastGI-23 lineage (Var2-like) of infectious bronchitis virus in Europe. Virus Res. 2017, 242, 43–48. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.M.; Jackwood, M.W.; Hilt, D.A. Identification of amino acids involved in a serotype and neutralization specific epitope within the S1 subunit of avian infectious bronchitis virus. Arch. Virol. 1997, 142, 2249–2256. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Huang, Y.C. Relationship between serotypes and genotypes based on the hypervariable region of the S1 gene of infectious bronchitis virus. Arch. Virol. 2000, 145, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Rohaim, M.A.; El Naggar, R.F.; Helal, A.M.; Bayoumi, M.; El-Saied, M.A.; Ahmed, K.A.; Shabbir, M.Z.; Munir, M. Genetic Diversity and Phylodynamics of Avian Coronaviruses in Egyptian Wild Birds. Viruses 2019, 11, 57. [Google Scholar] [CrossRef] [Green Version]
- Rohaim, M.A.; El Naggar, R.F.; Hamoud, M.M.; Bazid, A.I.; Gamal, A.M.; Laban, S.E.; Abdel-Sabour, M.A.; Nasr, S.A.E.; Zaki, M.M.; Shabbir, M.A.; et al. Emergence and genetic analysis of variant pathogenic 4/91 (serotype 793/B) infectious bronchitis virus in Egypt during 2019. Virus Genes 2019, 55, 720–725. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar]
- Zwaagstra, K.A.; Van Derzeijst, B.A.M.; Kusters, J.G. Rapid detection and identification of avian infectious bronchitis virus. J. Clin. Microbiol. 1992, 30, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Ababneh, M.; Dalab, A.E.; Alsaad, S.; Al-Zghoul, M. Presence of infectious bronchitis virus strain CK/CH/LDL/97I in the Middle East. ISRN Vet. Sci. 2012, 201721–201726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemida, M.G.; Al-Hammadi, M.A.; Daleb, A.H.S.; Gonsalves, C.R. Molecular characterization and phylogenetic analyses of virulent infectious bronchitis viruses isolated from chickens in Eastern Saudi Arabia. Virus Dis. 2017, 28, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Alsultan, M.A.; Alhammadi, M.A.; Hemida, M.G. Infectious bronchitis virus from chickens in Al-Hasa, Saudi Arabia 2015–2016. Vet. World 2019, 12, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Al-Mubarak, A.I.A.; Al-Kubati, A.A.G. Cocirculation of Four Infectious Bronchitis Virus Lineages in Broiler Chickens in the Eastern Region of Saudi Arabia from 2012 to 2014. Vet. Med. Int. 2020, 2020, 6037893–6037902. [Google Scholar] [CrossRef] [PubMed]
- Hussein, A.H.; Emara, M.M.; Rohaim, M.A. Sequence analysis of infectious bronchitis virus IS/1494 like strain isolated from broiler chicken co-infected with Newcastle disease virus in Egypt during 2012. Int. J. Poult. Sci. 2014, 13, 530–536. [Google Scholar] [CrossRef]
- Kahya, S.; Coven, F.; Temelli, S.; Eyigor, A.; Carli, K.T. Presence of IS/1494/06 genotype-related infectious bronchitis virus in breeder and broiler flocks in Turkey. Ankara Univ. Vet. Fak. Derg. 2013, 60, 27–31. [Google Scholar]
- Gandon, S.; Mackinnon, M.J.; Nee, S.; Read, A.F. Imperfect vaccines and the evolution of pathogen virulence. Nature 2001, 414, 751–775. [Google Scholar] [CrossRef] [Green Version]
- Franzo, G.; Legnardi, M.; Tucciarone, C.M.; Drigo, M.; Martini, M.; Cecchinato, M. Evolution of infectious bronchitis virus in the field after homologous vaccination introduction. Vet. Res. 2019, 50, 92. [Google Scholar]
- Read, A.F.; Baigent, S.J.; Powers, C.; Kgosana, L.B.; Blackwell, L.; Smith, L.P.; Kennedy, D.A.; Walkden-Brown, S.W.; Nair, V.K. Imperfect vaccination can enhance the transmission of highly virulent pathogens. PLoS Biol. 2015, 13, e1002198. [Google Scholar] [CrossRef]
- Xu, L.; Ren, M.; Sheng, J.; Ma, T.; Han, Z.; Zhao, Y.; Sun, J.; Liu, S. Genetic and biological characteristics of four novel recombinant avian infectious bronchitis viruses isolated in China. Virus Res. 2019, 263, 87–97. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Site | Sampled (n) | Breed | Positive (n) | Rate (%) |
|---|---|---|---|---|
| Buradiya | 7 | Broilers | 1 | 22.2 |
| 11 | Layers | 3 | ||
| Al Hofuf | 10 | Broilers | 1 | 12.5 |
| 6 | Layers | 1 | ||
| Dammam | 8 | Broilers | 2 | 15.4 |
| 5 | Layers | 0 | ||
| Al Duwadimi | 5 | Broilers | 0 | 9 |
| 6 | Layers | 1 |
| Strain | HVR1 (60–88) | HVR2 (115–140) | HVR3 (275–292) |
|---|---|---|---|
| 4/91 | VSVSDCTAGTFYESYNISAASVAMTVPPA | FKSQQGSCPLTGMIPQNHIRISAMRS | TFTNVSNASPNSGGVDTF |
| CR88 | G. A . . . . . . . . . . . H. . . . S . . . . . . HN | . . N . L . . . . . . . . . . . . . . . . . . . . D | S . . . . . . . . . . . . . . . . . |
| SA/1/2019 | . . AG . . . . . S . . . . K . . . . . . . . . . . . . . | . . . . . . . . . . . . . . . . . . . . . . . . . Y | . . . . . . . . . . . . . . . . . . |
| SA/2/2019 | . . AG . . . . . . . . . . . . F . . . . . . . . A . . . | . . . . . . . . . . . L . . . . . . . . . . . . . . | . . . . . . . . . . . . . . . . . . |
| SA/3/2019 | . . AGE . . . . . . . . . . . F . . S . . . . . A . . . | . . . . . . . . . . . . . . . . . . . . . . . . . Y | . . . . . . . . . . . . . . . . . . |
| SA/4/2019 | . . AGQ . . . . S. . . . . . . . . . . . . . . . . . | . . . . . . . . . . . . . . . . . . . . . . . . . . | . . . . . . . . . . . . . . . . . . |
| Strain | HVR1 (60–88) | HVR2 (115–140) | HVR3 (275–292) |
|---|---|---|---|
| IS/720/99 | Q- - IQCTVGAIGWSKNFSAASVAITAPAA | YSSGQGSCPLTGQLQRNSIRISAMSG | TFYNESNAPPNVGGVNTI |
| IS/1494/06 | G. . GQ . . A . S . Y . . . . . . . S . . . M . . . DT | . K . . H . . . . . . . LIPQ . H . . . . . . KN | . . T . V . . . S . . T . . . . . . |
| SA/5/2019 | G . . GQ . . A . S . Y . . . . . T . S . . . M . . . DT | . KN . . . . . . . . . LIPQ . H . . . . . . KN | . . T . V . . . S . . T . . . . . . |
| SA/6/2019 | G . . QQ . . A . . . Y . . . . . . . . . . . M . . . QN | . K . SS . . . . . . . MIPQYY . . . . . . RN | . . H . . . . . H . . N . . . H . . |
| SA/7/2019 | G . . QQ . . A . . . Y . . . . . . . . . . . M . . . QN | . K . SS . . . . . . . MIPQHY . . . . . . RN | . . . . . . . . S . . S . . . . . . |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rohaim, M.A.; El Naggar, R.F.; Abdelsabour, M.A.; Mohamed, M.H.A.; El-Sabagh, I.M.; Munir, M. Evolutionary Analysis of Infectious Bronchitis Virus Reveals Marked Genetic Diversity and Recombination Events. Genes 2020, 11, 605. https://doi.org/10.3390/genes11060605
Rohaim MA, El Naggar RF, Abdelsabour MA, Mohamed MHA, El-Sabagh IM, Munir M. Evolutionary Analysis of Infectious Bronchitis Virus Reveals Marked Genetic Diversity and Recombination Events. Genes. 2020; 11(6):605. https://doi.org/10.3390/genes11060605
Chicago/Turabian StyleRohaim, Mohammed A., Rania F. El Naggar, Mohammed A. Abdelsabour, Mahmoud H. A. Mohamed, Ibrahim M. El-Sabagh, and Muhammad Munir. 2020. "Evolutionary Analysis of Infectious Bronchitis Virus Reveals Marked Genetic Diversity and Recombination Events" Genes 11, no. 6: 605. https://doi.org/10.3390/genes11060605
APA StyleRohaim, M. A., El Naggar, R. F., Abdelsabour, M. A., Mohamed, M. H. A., El-Sabagh, I. M., & Munir, M. (2020). Evolutionary Analysis of Infectious Bronchitis Virus Reveals Marked Genetic Diversity and Recombination Events. Genes, 11(6), 605. https://doi.org/10.3390/genes11060605