Consequences of 22q11.2 Microdeletion on the Genome, Individual and Population Levels

Abstract
:
1. Introduction
2. Genome Level
2.1. Genotype-Specific Penetrance
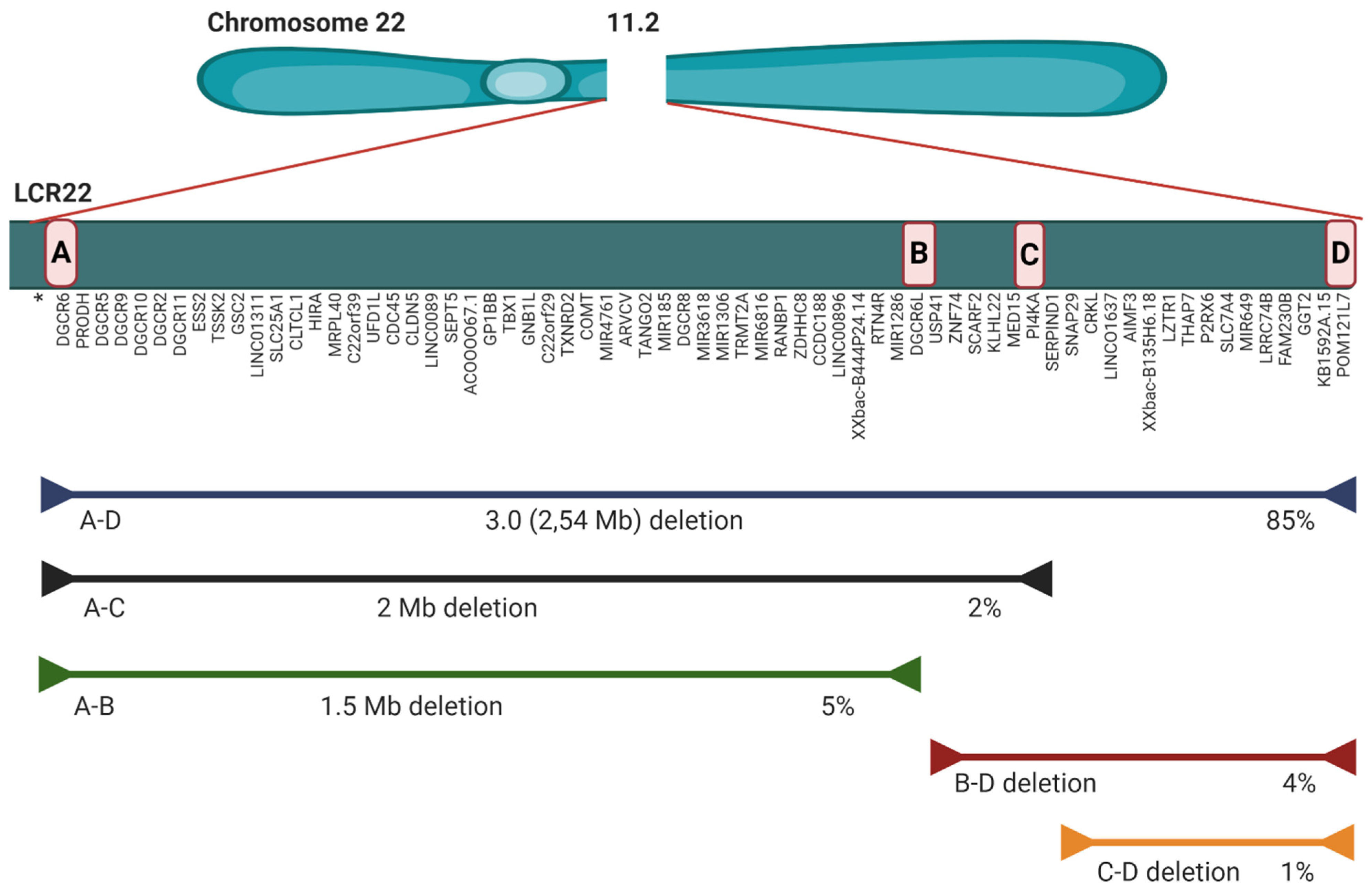
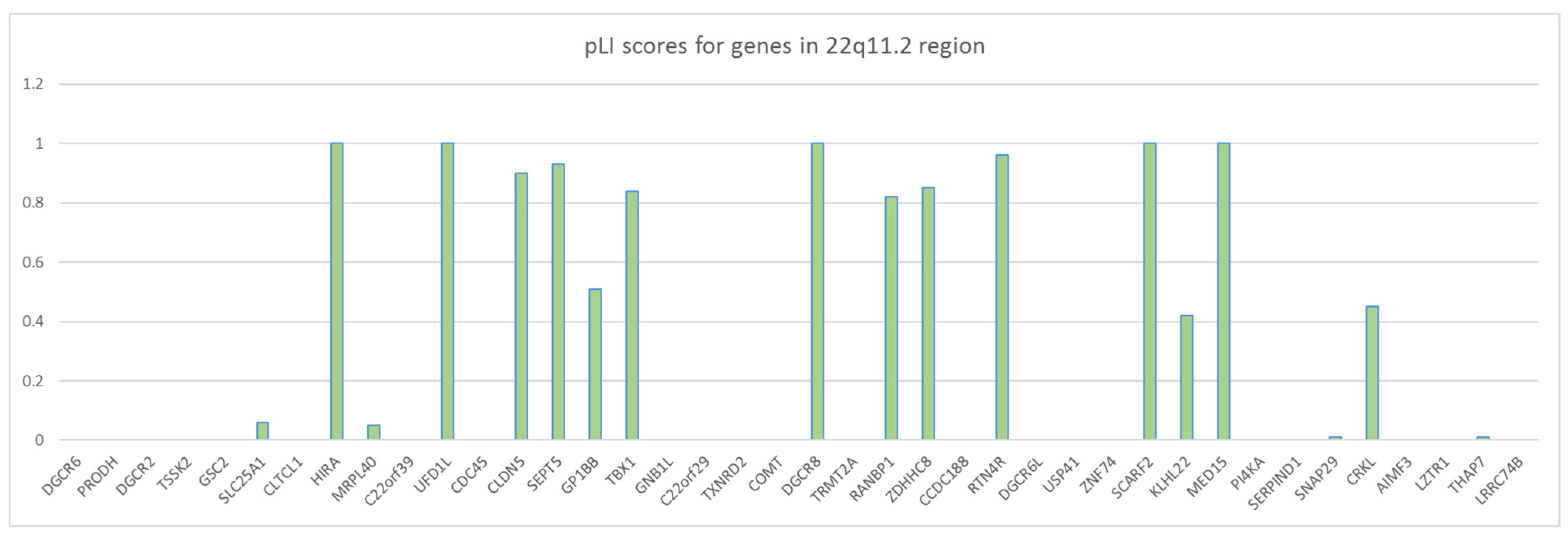
2.2. Microdeletion and Surroundings
2.3. Mouse Model of the 22q11.2 Deletion Syndrome
3. Individual Level
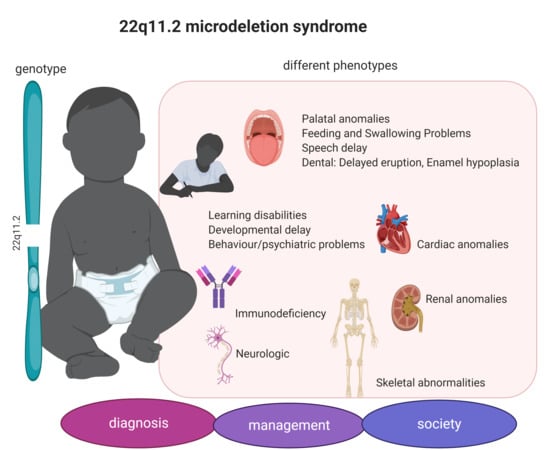
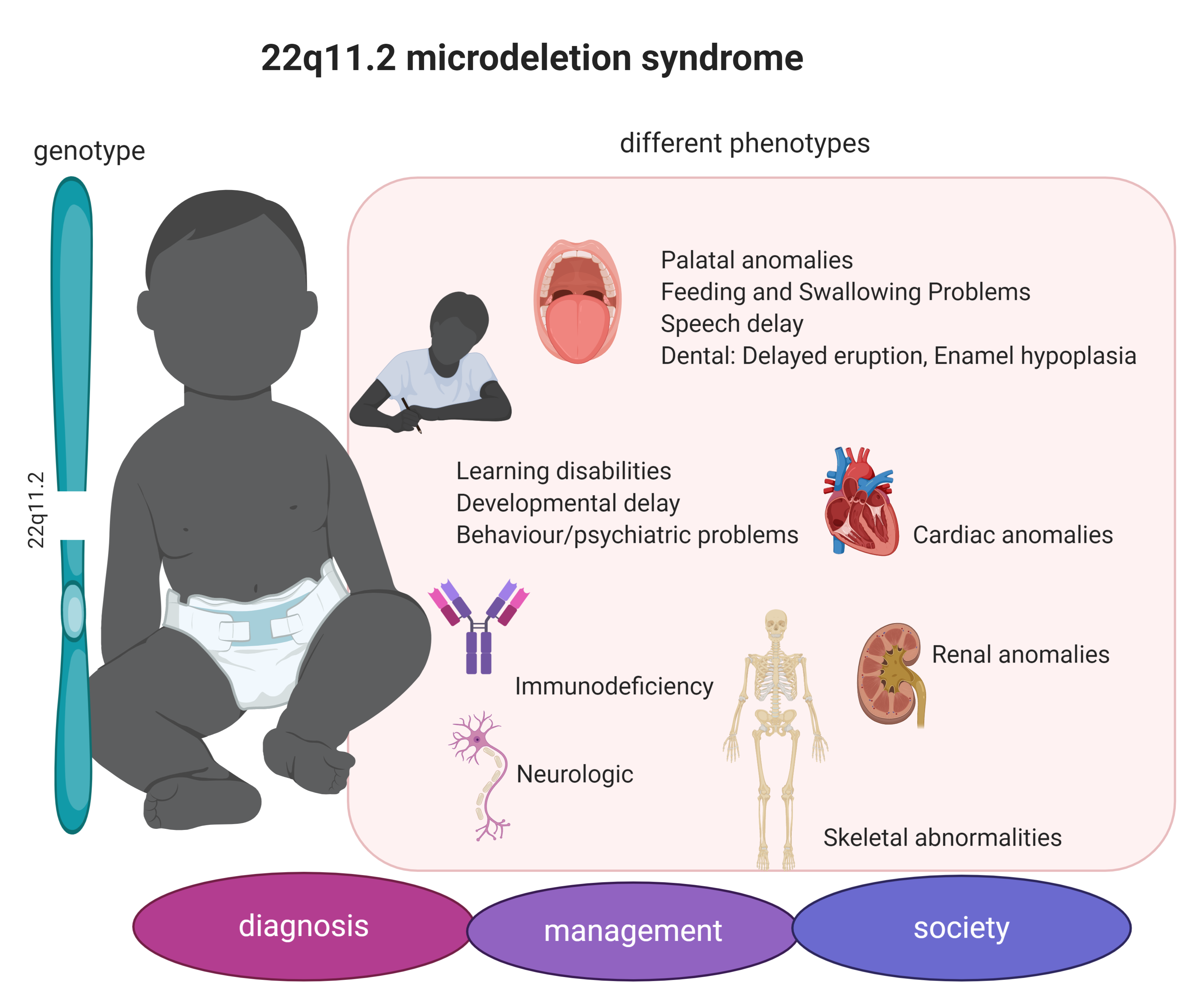
3.1. 22q11.2DS Phenotype
3.2. Diagnosis
4. Population Level
4.1. 22q11.2DS in Different Populations
4.2. Dealing with the Syndrome
5. Summary
Funding
Conflicts of Interest
References
- Wapner, R.J.; Martin, C.L.; Levy, B.; Ballif, B.C.; Eng, C.M.; Zachary, J.M.; Savage, M.; Platt, L.D.; Saltzman, D.; Grobman, W.A.; et al. Chromosomal Microarray versus Karyotyping for Prenatal Diagnosis. N. Engl. J. Med. 2012, 367, 2175–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grati, F.R.; Gomes, D.M.; Ferreira, J.; Dupont, C.; Alesi, V.; Gouas, L.; Horelli-Kuitunen, N.; Choy, K.W.; García-Herrero, S.; De La Vega, A.G.; et al. Prevalence of recurrent pathogenic microdeletions and microduplications in over 9500 pregnancies. Prenat. Diagn. 2015, 35, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Fung, W.L.A.; Butcher, N.J.; Costain, G.; Andrade, D.M.; Boot, E.; Chow, E.W.; Chung, B.; Cytrynbaum, C.; Faghfoury, H.; Fishman, L.; et al. Practical guidelines for managing adults with 22q11.2 deletion syndrome. Genet. Med. 2015, 17, 599–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald-McGinn, D.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.S.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E.; et al. 22q11.2 deletion syndrome. Nat. Rev. Dis. Primers 2015, 1, 15071. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, K.E. Chromosome 22q11.2 deletion syndrome and DiGeorge syndrome. Immunol. Rev. 2018, 287, 186–201. [Google Scholar] [CrossRef]
- Botto, L.D.; May, K.; Fernhoff, P.M.; Correa, A.; Coleman, K.; Rasmussen, S.A.; Merritt, R.K.; O’Leary, L.A.; Wong, L.-Y.; Elixson, E.M.; et al. A population-based study of the 22q11.2 deletion: Phenotype, incidence, and contribution to major birth defects in the population. Pediatrics 2003, 112, 101–107. [Google Scholar] [CrossRef]
- Motahari, Z.; Moody, S.A.; Maynard, T.M.; LaMantia, A.-S. In the line-up: Deleted genes associated with DiGeorge/22q11.2 deletion syndrome: Are they all suspects? J. Neurodev. Disord. 2019, 11, 7. [Google Scholar] [CrossRef] [Green Version]
- Michaelovsky, E.; Frisch, A.; Carmel, M.; Patya, M.; Zarchi, O.; Green, T.; Basel-Vanagaite, V.L.; Weizman, A.; Gothelf, D. Genotype-phenotype correlation in 22q11.2 deletion syndrome. BMC Med. Genet. 2012, 13, 122. [Google Scholar] [CrossRef] [Green Version]
- Sandrin-Garcia, P.; Abramides, D.V.M.; Martelli, L.R.; Ramos, E.S.; Richieri-Costa, A.; Passos, G.A. Typical phenotypic spectrum of velocardiofacial syndrome occurs independently of deletion size in chromosome 22q11. Mol. Cell. Biochem. 2007, 303, 9–17. [Google Scholar] [CrossRef]
- Saitta, S.C.; Harris, S.E.; Gaeth, A.P.; Driscoll, D.A.; McDonald-McGinn, D.M.; Maisenbacher, M.K.; Yersak, J.M.; Chakraborty, P.K.; Hacker, A.M.; Zackai, E.H.; et al. Aberrant interchromosomal exchanges are the predominant cause of the 22q11.2 deletion. Hum. Mol. Genet. 2003, 13, 417–428. [Google Scholar] [CrossRef]
- Babcock, M.; Pavlicek, A.; Spiteri, E.; Kashork, C.D.; Ioshikhes, I.; Shaffer, L.G.; Jurka, J.; Morrow, B. Shuffling of Genes Within Low-Copy Repeats on 22q11 (LCR22) by Alu-Mediated Recombination Events During Evolution. Genome Res. 2003, 13, 2519–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bittel, D.C.; Yu, S.; Newkirk, H.; Kibiryeva, N.; Holt, A.; Butler, M.; Cooley, L.; Holt, S. Refining the 22q11.2 deletion breakpoints in DiGeorge syndrome by aCGH. Cytogenet. Genome Res. 2009, 124, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Lupski, J.R. MOLECULARMECHANISMS FORGENOMICDISORDERS. Annu. Rev. Genom. Hum. Genet. 2002, 3, 199–242. [Google Scholar] [CrossRef] [PubMed]
- Weise, A.; Mrasek, K.; Klein, E.; Mulatinho, M.; Llerena, J.C.; Hardekopf, D.; Pekova, S.; Bhatt, S.; Kosyakova, N.; Liehr, T. Microdeletion and Microduplication Syndromes. J. Histochem. Cytochem. 2012, 60, 346–358. [Google Scholar] [CrossRef] [Green Version]
- Portnoi, M.-F. Microduplication 22q11.2: A new chromosomal syndrome. Eur. J. Med. Genet. 2009, 52, 88–93. [Google Scholar] [CrossRef]
- 22q11.2 Duplication. Available online: https://www.ncbi.nlm.nih.gov/pubmed/ (accessed on 25 July 2020).
- Unolt, M.; Versacci, P.; Anaclerio, S.; Lambiase, C.; Calcagni, G.; Trezzi, M.; Carotti, A.; Crowley, T.B.; Zackai, E.H.; Goldmuntz, E.; et al. Congenital heart diseases and cardiovascular abnormalities in 22q11.2 deletion syndrome: From well-established knowledge to new frontiers. Am. J. Med. Genet. Part A 2018, 176, 2087–2098. [Google Scholar] [CrossRef]
- Du, Q.; De La Morena, M.T.; Van Oers, N.S.C. The Genetics and Epigenetics of 22q11.2 Deletion Syndrome. Front. Genet. 2020, 10. [Google Scholar] [CrossRef]
- Dyce, O.; McDonald-McGinn, N.; Kirschner, R.E.; Zackai, E.; Young, K.; Jacobs, I.N. Otolaryngologic manifestations of the 22q11.2 deletion syndrome. Arch. Otolaryngol. Head Neck Surg. 2002, 128, 1408–1412. [Google Scholar] [CrossRef] [Green Version]
- De Smedt, B.; Swillen, A.; Verschaffel, L.; Ghesquiere, P. Mathematical learning disabilities in children with 22q11.2 deletion syndrome: A review. Dev. Disabil. Res. Rev. 2009, 15, 4–10. [Google Scholar] [CrossRef]
- Weinzimer, S.A. Endocrine aspects of the 22q11.2 deletion syndrome. Genet. Med. 2001, 3, 19–22. [Google Scholar] [CrossRef] [Green Version]
- Goldmuntz, E. 22q11.2 deletion syndrome and congenital heart disease. Am. J. Med. Genet. Part C Semin. Med. Genet. 2020, 184, 64–72. [Google Scholar] [CrossRef] [PubMed]
- McDonald-McGinn, D.; Tonnesen, M.K.; Laufer-Cahana, A.; Finucane, B.; A Driscoll, D.; Emanuel, B.S.; Zackai, E.H. Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: Cast a wide FISHing net! Genet. Med. 2001, 3, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, H.; Ishii, C.; Maeda, J.; Kojima, Y.; Matsuoka, R.; Kimura, M.; Takao, A.; Momma, K.; Matsuo, N. Phenotypic discordance in monozygotic twins with 22q11.2 deletion. Am. J. Med. Genet. 1998, 78, 319–321. [Google Scholar] [CrossRef]
- Fernandez, L.; Nevado, J.; Santos, F.; Heine-Suñer, D.; Martínez-Glez, V.; García-Miñaur, S.; Palomo, R.; Delicado, A.; Pajares, I.L.; Bralo, M.P.; et al. A deletion and a duplication in distal 22q11.2 deletion syndrome region. Clinical implications and review. BMC Med. Genet. 2009, 10, 48. [Google Scholar] [CrossRef] [Green Version]
- A Driscoll, D.; Salvin, J.; Sellinger, B.; Budarf, M.L.; McDonald-McGinn, D.M.; Zackai, E.H.; Emanuel, B.S. Prevalence of 22q11 microdeletions in DiGeorge and velocardiofacial syndromes: Implications for genetic counselling and prenatal diagnosis. J. Med. Genet. 1993, 30, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Conrad, D.F.; The Wellcome Trust Case Control Consortium; Pinto, D.; Redon, R.; Feuk, L.; Gokcumen, O.; Zhang, Y.; Aerts, J.; Andrews, T.D.; Barnes, C.P.; et al. Origins and functional impact of copy number variation in the human genome. Nature 2009, 464, 704–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The 1000 Genomes Project Consortium; 1000 Genomes Project Consortium; Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A.; et al. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, D.A.; Srinivasan, M.; Egholm, M.; Shen, Y.; Chen, L.; McGuire, A.; He, W.; Chen, Y.-J.; Makhijani, V.; Roth, G.T.; et al. The complete genome of an individual by massively parallel DNA sequencing. Nature 2008, 452, 872–876. [Google Scholar] [CrossRef]
- Abel, H.J.; Genomics, N.C.F.C.D.; Larson, D.E.; Regier, A.A.; Chiang, C.; Das, I.; Kanchi, K.L.; Layer, R.M.; Neale, B.M.; Salerno, W.J.; et al. Mapping and characterization of structural variation in 17,795 human genomes. Nature 2020, 583, 83–89. [Google Scholar] [CrossRef]
- Collins, R.L.; Brand, H.; Karczewski, K.; Zhao, X.; Alföldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef]
- Cooper, D.N.; Krawczak, M.; Polychronakos, C.; Tyler-Smith, C.; Kehrer-Sawatzki, H. Where genotype is not predictive of phenotype: Towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Qual. Life Res. 2013, 132, 1077–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guna, A.; Butcher, N.J.; Bassett, A.S. Comparative mapping of the 22q11.2 deletion region and the potential of simple model organisms. J. Neurodev. Disord. 2015, 7, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertini, V.; Azzarà, A.; Legitimo, A.; Milone, R.; Battini, R.; Consolini, R.; Valetto, A. Deletion Extents Are Not the Cause of Clinical Variability in 22q11.2 Deletion Syndrome: Does the Interaction between DGCR8 and miRNA-CNVs Play a Major Role? Front. Genet. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastor, S.; Tran, O.; Jin, A.; Carrado, D.; Silva, B.A.; Uppuluri, L.; Abid, H.Z.; Young, E.; Crowley, T.B.; Bailey, A.G.; et al. Optical mapping of the 22q11.2DS region reveals complex repeat structures and preferred locations for non-allelic homologous recombination (NAHR). Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.; Ware, J.S.; Hill, A.J.; Cummings, B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GnomAD. Available online: https://gnomad.broadinstitute.org/ (accessed on 23 July 2020).
- Fulcoli, F.G.; Franzese, M.; Liu, X.; Zhang, Z.; Angelini, C.; Baldini, A. Rebalancing gene haploinsufficiency in vivo by targeting chromatin. Nat. Commun. 2016, 7, 11688. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Baldini, A. In vivo response to high-resolution variation of Tbx1 mRNA dosage. Hum. Mol. Genet. 2007, 17, 150–157. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, D.; Bernal, A.J.; Schoch, K.; Howard, T.D.; Ip, E.H.; Hooper, S.R.; Keshavan, M.S.; Jirtle, R.L.; Shashi, V.; Das Chakraborty, R. Dysregulation of DGCR6 and DGCR6L: Psychopathological outcomes in chromosome 22q11.2 deletion syndrome. Transl. Psychiatry 2012, 2, e105. [Google Scholar] [CrossRef] [Green Version]
- De La Morena, M.T.; Eitson, J.L.; Dozmorov, I.M.; Belkaya, S.; Hoover, A.R.; Anguiano, E.; Pascual, M.V.; Van Oers, N.S.C. Signature MicroRNA expression patterns identified in humans with 22q11.2 deletion/DiGeorge syndrome. Clin. Immunol. 2013, 147, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Sellier, C.; Hwang, V.J.; Dandekar, R.; Durbin-Johnson, B.; Charlet-Berguerand, N.; Ander, B.P.; Sharp, F.R.; Angkustsiri, K.; Simon, T.J.; Tassone, F. Decreased DGCR8 Expression and miRNA Dysregulation in Individuals with 22q11.2 Deletion Syndrome. PLoS ONE 2014, 9, e103884. [Google Scholar] [CrossRef]
- Barr, I.; Weitz, S.H.; Atkin, T.; Hsu, P.; Karayiorgou, M.; Gogos, J.A.; Weiss, S.; Guo, F. Cobalt(III) Protoporphyrin Activates the DGCR8 Protein and Can Compensate microRNA Processing Deficiency. Chem. Biol. 2015, 22, 793–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- León, L.E.; Benavides, F.; Espinoza, K.; Vial, C.; Alvarez, P.; Palomares, M.; Lay-Son, G.; Miranda, M.; Repetto, G.M. Partial microduplication in the histone acetyltransferase complex member KANSL1 is associated with congenital heart defects in 22q11.2 microdeletion syndrome patients. Sci. Rep. 2017, 7, 1795. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Chung, J.; Wang, T.; McDonald-McGinn, D.M.; Kates, W.R.; Hawuła, W.; Coleman, K.; Zackai, E.; Emanuel, B.S.; Morrow, B.E. Histone modifier genes alter conotruncal heart phenotypes in 22q11.2 deletion syndrome. Am. J. Hum. Genet. 2015, 97, 869–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mlynarski, E.E.; Sheridan, M.B.; Xie, M.; Guo, T.; Racedo, S.E.; McDonald-McGinn, D.M.; Gai, X.; Chow, E.W.; Vorstman, J.; Swillen, A.; et al. Copy-Number Variation of the Glucose Transporter Gene SLC2A3 and Congenital Heart Defects in the 22q11.2 Deletion Syndrome. Am. J. Hum. Genet. 2015, 96, 753–764. [Google Scholar] [CrossRef] [Green Version]
- Hiramoto, T.; Kang, G.; Suzuki, G.; Satoh, Y.; Kucherlapati, R.; Watanabe, Y.; Hiroi, N. Tbx1: Identification of a 22q11.2 gene as a risk factor for autism spectrum disorder in a mouse model. Hum. Mol. Genet. 2011, 20, 4775–4785. [Google Scholar] [CrossRef] [Green Version]
- Stark, K.L.; Xu, B.; Bagchi, A.; Lai, W.-S.; Liu, H.; Hsu, R.; Wan, X.; Pavlidis, P.; Mills, A.A.; Karayiorgou, M.; et al. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 2008, 40, 751–760. [Google Scholar] [CrossRef]
- Ouchi, Y.; Banno, Y.; Shimizu, Y.; Ando, S.; Hasegawa, H.; Adachi, K.; Iwamoto, T. Reduced Adult Hippocampal Neurogenesis and Working Memory Deficits in the Dgcr8-Deficient Mouse Model of 22q11.2 Deletion-Associated Schizophrenia Can Be Rescued by IGF2. J. Neurosci. 2013, 33, 9408–9419. [Google Scholar] [CrossRef]
- Nilsson, S.R.; Fejgin, K.; Gastambide, F.; Vogt, M.A.; A Kent, B.; Nielsen, V.; Nielsen, J.; Gass, P.; Robbins, T.W.; Saksida, L.M.; et al. Assessing the Cognitive Translational Potential of a Mouse Model of the 22q11.2 Microdeletion Syndrome. Cereb. Cortex 2016, 26, 3991–4003. [Google Scholar] [CrossRef] [Green Version]
- Saito, R.; Koebis, M.; Nagai, T.; Shimizu, K.; Liao, J.; Wulaer, B.; Sugaya, Y.; Nagahama, K.; Uesaka, N.; Kushima, I.; et al. Comprehensive analysis of a novel mouse model of the 22q11.2 deletion syndrome: A model with the most common 3.0-Mb deletion at the human 22q11.2 locus. Transl. Psychiatry 2020, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Gennery, A.R. Immunological aspects of 22q11.2 deletion syndrome. Cell. Mol. Life Sci. 2011, 69, 17–27. [Google Scholar] [CrossRef]
- Morsheimer, M.; Brown-Whitehorn, T.; Heimall, J.; Sullivan, K.E. The immune deficiency of chromosome 22q11.2 deletion syndrome. Am. J. Med. Genet. Part A 2017, 173, 2366–2372. [Google Scholar] [CrossRef]
- Ziółkowska, L.; Kawalec, W.; Turska-Kmieć, A.; Krajewska-Walasek, M.; Brzezińska-Rajszys, G.; Daszkowska, J.; Maruszewski, B.; Burczynski, P.L. Chromosome 22q11.2 microdeletion in children with conotruncal heart defects: Frequency, associated cardiovascular anomalies, and outcome following cardiac surgery. Eur. J. Nucl. Med. Mol. Imaging 2008, 167, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Goldmuntz, E.; Clark, B.J.; E Mitchell, L.; Jawad, A.F.; Cuneo, B.F.; Reed, L.; McDonald-McGinn, N.; Chien, P.; Feuer, J.; Zackai, E.H.; et al. Frequency of 22q11 deletions in patients with conotruncal defects. J. Am. Coll. Cardiol. 1998, 32, 492–498. [Google Scholar] [CrossRef] [Green Version]
- Kauw, D.; Woudstra, O.I.; Van Engelen, K.; Meijboom, F.J.; Mulder, B.J.; Schuuring, M.J.; Bouma, B. 22q11.2 deletion syndrome is associated with increased mortality in adults with tetralogy of Fallot and pulmonary atresia with ventricular septal defect. Int. J. Cardiol. 2020, 306, 56–60. [Google Scholar] [CrossRef]
- Wu, H.Y.; Rusnack, S.L.; Bellah, R.D.; Plachter, N.; McDonald-McGinn, D.M.; Zackai, E.H.; Canning, D.A. Genitourinary Malformations in Chromosome 22q11.2 Deletion. J. Urol. 2002, 168, 2564–2565. [Google Scholar] [CrossRef]
- Eicher, P.S.; McDonald-McGinn, D.M.; Fox, C.A.; Driscoll, D.A.; Emanuel, B.S.; Zackai, E.H. Dysphagia in children with a 22q11.2 deletion: Unusual pattern found on modified barium swallow. J. Pediatr. 2000, 137, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Welby, L.; Caudill, H.; Yitsege, G.; Hamad, A.; Bunyak, F.; Zohn, I.E.; Maynard, T.; LaMantia, A.-S.; Mendelowitz, D.; Lever, T.E. Persistent Feeding and Swallowing Deficits in a Mouse Model of 22q11.2 Deletion Syndrome. Front. Neurol. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Wozniak, A.; Wolnik-Brzozowska, D.; Wisniewska, M.; Glazar, R.; Materna-Kiryluk, A.; Moszura, T.; Badura-Stronka, M.; Skolozdrzy, J.; Krawczyński, M.R.; Zeyland, J.; et al. Frequency of 22q11.2 microdeletion in children with congenital heart defects in western poland. BMC Pediatr. 2010, 10, 88. [Google Scholar] [CrossRef] [Green Version]
- Koehler, S.; Carmody, L.C.; Vasilevsky, N.; Jacobsen, J.O.B.; Danis, D.; Gourdine, J.-P.; Gargano, M.; Harris, N.L.; Matentzoglu, N.; A McMurry, J.; et al. Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res. 2018, 47, D1018–D1027. [Google Scholar] [CrossRef]
- Murphy, K.C.; A Jones, L.; Owen, M.J. High rates of schizophrenia in adults with velo-cardio-facial syndrome. Arch. Gen. Psychiatry 1999, 56, 940–945. [Google Scholar] [CrossRef] [Green Version]
- Bassett, A.S.; Chow, E.W.; Abdelmalik, P.; Gheorghiu, M.; Husted, J.; Weksberg, R. The schizophrenia phenotype in 22q11 deletion syndrome. Am. J. Psychiatry 2003, 160, 1580–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleynen, I.; Engchuan, W.; Hestand, M.S.; Heung, T.; Holleman, A.M.; Johnston, H.R.; Monfeuga, T.; McDonald-McGinn, D.; Gur, R.E.; Morrow, B.E.; et al. Genetic contributors to risk of schizophrenia in the presence of a 22q11.2 deletion. Mol. Psychiatry 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Vajdi, A.; Kushan-Wells, L.; Hellemann, G.; Hansen, L.P.; Jonas, R.K.; Jalbrzikowski, M.; Kingsbury, L.; Raznahan, A.; Bearden, C.E. Reciprocal Copy Number Variations at 22q11.2 Produce Distinct and Convergent Neurobehavioral Impairments Relevant for Schizophrenia and Autism Spectrum Disorder. Biol. Psychiatry 2020. [Google Scholar] [CrossRef] [PubMed]
- Armando, M.; Ciampoli, M.; Padula, M.C.; Amminger, P.; De Crescenzo, F.; Maeder, J.; Schneider, M.; Schaer, M.; Manago, F.; Eliez, S.; et al. Favorable effects of omega-3 polyunsaturated fatty acids in attentional control and conversion rate to psychosis in 22q11.2 deletion syndrome. Neuropharmacology 2020, 168, 107995. [Google Scholar] [CrossRef] [PubMed]
- Sandini, C.; Chambaz, M.; Schneider, M.; Armando, M.; Zöller, D.; Schaer, M.; Sandi, C.; Van De Ville, D.; Eliez, S. Pituitary dysmaturation affects psychopathology and neurodevelopment in 22q11.2 Deletion Syndrome. Psychoneuroendocrinology 2020, 113, 104540. [Google Scholar] [CrossRef] [PubMed]
- Moulding, H.A.; Bartsch, U.; Hall, J.; Jones, M.W.; Linden, D.E.; Owen, M.J.; Bree, M.B.M. Sleep problems and associations with psychopathology and cognition in young people with 22q11.2 deletion syndrome (22q11.2DS). Psychol. Med. 2019, 50, 1191–1202. [Google Scholar] [CrossRef] [Green Version]
- Gerdes, M.; Solot, C.; Wang, P.P.; Moss, E.; LaRossa, D.; Randall, P.; Goldmuntz, E.; Clark, B.J.; Driscoll, D.A.; Jawad, A.; et al. Cognitive and Behavior Profile of Preschool Children With Chromosome 22q11.2 Deletion. Am. J. Med Genet. 1999, 85, 127–133. [Google Scholar] [CrossRef]
- Gerdes, M.; Solot, C.; Wang, P.P.; McDonald-McGinn, D.; Zackai, E.H. Taking advantage of early diagnosis: Preschool children with the 22q11.2 deletion. Genet. Med. 2001, 3, 40–44. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, A.C.; Fung, W.; Massey, T.H.; Hall, J.; Owen, M.J.; Bree, M.B.M.; Peall, K.J.; Massey, T.H.; Owen, M.J.; Peall, K.J. Movement Disorder Phenotypes in Children With 22q11.2 Deletion Syndrome. Mov. Disord. 2020, 35, 1272–1274. [Google Scholar] [CrossRef]
- Solot, C.B.; Gerdes, M.; Kirschner, R.E.; McDonald-McGinn, D.; Moss, E.; Woodin, M.; Aleman, D.; Zackai, E.H.; Wang, P.P. Communication issues in 22q11.2 deletion syndrome: Children at risk. Genet. Med. 2001, 3, 67–71. [Google Scholar] [CrossRef] [Green Version]
- Swillen, A. The importance of understanding cognitive trajectories: The case of 22q11.2 deletion syndrome. Curr. Opin. Psychiatry 2016, 29, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Solot, C.B.; Sell, D.; Mayne, A.; Baylis, A.L.; Persson, C.; Jackson, O.; McDonald-McGinn, D.M. Speech-Language Disorders in 22q11.2 Deletion Syndrome: Best Practices for Diagnosis and Management. Am. J. Speech Lang. Pathol. 2019, 28, 984–999. [Google Scholar] [CrossRef] [Green Version]
- Campbell, I.M.; Sheppard, S.E.; Crowley, T.B.; McGinn, D.E.; Bailey, A.; McGinn, M.J.; Unolt, M.; Homans, J.F.; Chen, E.Y.; Salmons, H.I.; et al. What’s New with 22q? An update from the 22q and You Center at the Children’s Hospital of Philadelphia. Am. J. Med. Genet. Part A 2018, 176, 2058–2069. [Google Scholar] [CrossRef] [PubMed]
- Moss, E.M.; Batshaw, M.L.; Solot, C.B.; Gerdes, M.; McDonald-McGinn, D.; Driscoll, D.A.; Emanuel, B.S.; Zackai, E.H.; Wang, P.P. Psychoeducational profile of the 22q11.2 microdeletion: A complex pattern. J. Pediatr. 1999, 134, 193–198. [Google Scholar] [CrossRef]
- Lajiness-O’Neill1, R.R.; Beaulieu, I.; Titus, J.B.; Asamoah, A.; Bigler, E.D.; Bawle, E.V.; Pollack, R.; Lajiness-O’Neill, R. Memory and Learning in Children with 22q11.2 Deletion Syndrome: Evidence for Ventral and Dorsal Stream Disruption? Child. Neuropsychol. 2005, 11, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Schoch, K.; Harrell, W.; Hooper, S.R.; Ip, E.H.; Saldana, S.; Kwapil, T.R.; Shashi, V. Applicability of the Nonverbal Learning Disability Paradigm for Children With 22q11.2 Deletion Syndrome. J. Learn. Disabil. 2012, 47, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro-Bicudo, L.; Legnaro, C.D.C.; Gamba, B.; Sandri, R.C.; Richieri-Costa, A. Cognitive deficit, learning difficulties, severe behavioral abnormalities and healed cleft lip in a patient with a 1.2-mb distal microduplication at 22q11. Mol. Syndromol. 2013, 4, 292–296. [Google Scholar] [CrossRef]
- Shashi, V.; Veerapandiyan, A.; Schoch, K.; Kwapil, T.R.; Keshavan, M.; Ip, E.; Hooper, S. Social skills and associated psychopathology in children with chromosome 22q11.2 deletion syndrome: Implications for interventions. J. Intellect. Disabil. Res. 2011, 56, 865–878. [Google Scholar] [CrossRef]
- Briegel, W.; Schneider, M.; Schwab, K.O. 22q11.2 deletion syndrome: Behaviour problems of children and adolescents and parental stress. Child. Care Health Dev. 2008, 34, 34. [Google Scholar] [CrossRef]
- Campbell, L.E.; Stevens, A.F.; McCabe, K.; Cruickshank, L.; Morris, R.G.; Murphy, D.; Murphy, K.C. Is theory of mind related to social dysfunction and emotional problems in 22q11.2 deletion syndrome (velo-cardio-facial syndrome)? J. Neurodev. Disord. 2011, 3, 152–161. [Google Scholar] [CrossRef] [Green Version]
- Jalbrzikowski, M.; Carter, C.; Şentürk, D.; Chow, C.; Hopkins, J.M.; Green, M.F.; Galvan, A.; Cannon, T.D.; Bearden, C.E. Social cognition in 22q11.2 microdeletion syndrome: Relevance to psychosis? Schizophr. Res. 2012, 142, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angkustsiri, K.; Leckliter, I.; Tartaglia, N.; Beaton, E.A.; Enriquez, J.; Simon, T.J. An Examination of the Relationship of Anxiety and Intelligence to Adaptive Functioning in Children with Chromosome 22q11.2 Deletion Syndrome. J. Dev. Behav. Pediatr. 2012, 33, 713–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, F.; Hayiou-Thomas, M.E.; Arshad, Z.; Leonard, M.; Williams, F.J.; Katseniou, N.; Malouta, R.N.; Marshall, C.R.P.; Diamantopoulou, M.; Tang, E.; et al. Mind-Mindedness and Stress in Parents of Children with Developmental Disorders. J. Autism Dev. Disord. 2020, 1–13. [Google Scholar] [CrossRef]
- Swillen, A.; McDonald-McGinn, D.; McDonald-McGinn, D. Developmental trajectories in 22q11.2 deletion syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 2015, 169, 172–181. [Google Scholar] [CrossRef] [Green Version]
- Mosheva, M.; Pouillard, V.; Fishman, Y.; Dubourg, L.; Sofrin-Frumer, D.; Serur, Y.; Weizman, A.; Eliez, S.; Gothelf, R.; Schneider, M. Education and employment trajectories from childhood to adulthood in individuals with 22q11.2 deletion syndrome. Eur. Child. Adolesc. Psychiatry 2018, 28, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Van, L.; Heung, T.; Graffi, J.; Ng, E.; Malecki, S.; Van Mil, S.; Boot, E.; Corral, M.; Chow, E.W.C.; Hodgkinson, K.A.; et al. All-cause mortality and survival in adults with 22q11.2 deletion syndrome. Genet. Med. 2019, 21, 2328–2335. [Google Scholar] [CrossRef] [Green Version]
- Bretelle, F.; Beyer, L.; Pellissier, M.C.; Missirian, C.; Sigaudy, S.; Gamerre, M.; D’Ercole, C.; Philip, N. Prenatal and postnatal diagnosis of 22q11.2 deletion syndrome. Eur. J. Med. Genet. 2010, 53, 367–370. [Google Scholar] [CrossRef]
- Fernandez, L.; Lapunzina, P.; Arjona, D.; Pajares, I.L.; García-Guereta, L.; Elorza, D.; Burgueros, M.; De Torres, M.; Mori, M.; Palomares, M.; et al. Comparative study of three diagnostic approaches (FISH, STRs and MLPA) in 30 patients with 22q11.2 deletion syndrome. Clin. Genet. 2005, 68, 373–378. [Google Scholar] [CrossRef]
- Shefi, S.; Raviv, G.; Rienstein, S.; Barkai, G.; Aviram-Goldring, A.; Levron, J. Fish based preimplantation genetic diagnosis to prevent DiGeorge syndrome. J. Assist. Reprod. Genet. 2009, 26, 411–413. [Google Scholar] [CrossRef] [Green Version]
- Iwarsson, E.; Ahrlund-Richter, L.; Inzunza, J.; Fridström, M.; Rosenlund, B.; Hillensjö, T.; Sjöblom, P.; Nordenskjöld, M.; Blennow, E. Preimplantation genetic diagnosis of DiGeorge syndrome. Mol. Hum. Reprod. 1998, 4, 871–875. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Jin, Y.; Yang, J.; Yang, L.; Tang, P.; Zhou, C.; Wu, L.; Dong, J.; Chen, J.; Shen, H. Prenatal diagnosis of rearrangements in the fetal 22q11.2 region. Mol. Cytogenet. 2020, 13, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Dugoff, L.; Mennuti, M.T.; McDonald-McGinn, D.M. The benefits and limitations of cell-free DNA screening for 22q11.2 deletion syndrome. Prenat. Diagn. 2016, 37, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.J.; Dzakula, Z.; Deciu, C.; Boom, D.V.D.; Ehrich, M. Detection of Microdeletion 22q11.2 in a Fetus by Next-Generation Sequencing of Maternal Plasma. Clin. Chem. 2012, 58, 1148–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, S.J.; Stosic, M.; McDonald-McGinn, D.M.; Bassett, A.S.; Norvez, A.; Dhamankar, R.; Kobara, K.; Kirkizlar, E.; Zimmermann, B.; Wayham, N.; et al. Clinical experience with single-nucleotide polymorphism-based non-invasive prenatal screening for 22q11.2 deletion syndrome. Ultrasound Obstet. Gynecol. 2016, 47, 177–183. [Google Scholar] [CrossRef]
- Lewandowicz-Uszyńska, A.; Zwonarz, K.; Chmielarska, J. The 22q11 microdeletion syndrome in children. Central Eur. J. Immunol. 2013, 2, 271–275. [Google Scholar] [CrossRef] [Green Version]
- Halder, A.; Jain, M.; Kalsi, A.K. SNP Microarray in FISH Negative Clinically Suspected 22q11.2 Microdeletion Syndrome. Scientifica 2016, 2016, 1–18. [Google Scholar] [CrossRef] [Green Version]
- How Is Genetic Testing for 22q11.2 Deletions Done? Available online: https://22q.ca/medicalprofessionals/how-is-screening-done/#genomic-snp-array (accessed on 25 July 2020).
- Sandrin-Garcia, P.; Macedo, C.; Martelli, L.; Ramos, E.S.; Guion-Almeida, M.; Richieri-Costa, A.; Passos, G.A. Recurrent 22q11.2 deletion in a sibship suggestive of parental germline mosaicism in velocardiofacial syndrome. Clin. Genet. 2002, 61, 380–383. [Google Scholar] [CrossRef]
- Moreno-De-Luca, A.; Myers, S.M.; Challman, T.D.; Moreno-De-Luca, D.; Evans, D.W.; Ledbetter, D.H. Developmental brain dysfunction: Revival and expansion of old concepts based on new genetic evidence. Lancet Neurol. 2013, 12, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Kruszka, P.; Addissie, Y.A.; McGinn, D.E.; Porras, A.R.; Biggs, E.; Share, M.; Crowley, T.B.; Chung, B.H.Y.; Mok, G.T.K.; Mak, C.C.Y.; et al. 22q11.2 deletion syndrome in diverse populations. Am. J. Med. Genet. Part A 2017, 173, 879–888. [Google Scholar] [CrossRef]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Atlas of Human Malformation Syndromes in Diverse Populations. Available online: https://research.nhgri.nih.gov/atlas/ (accessed on 24 July 2020).
- Adani, S.; Cepanec, M. Sex differences in early communication development: Behavioral and neurobiological indicators of more vulnerable communication system development in boys. Croat. Med. J. 2019, 60, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, R.; Bree, M.V.D.; Challenger, A.; Cuthbert, A.; Ayllon, M.A.; Clarke, A.; Thompson, R. Co-creating a knowledge base in the “22q11.2 deletion syndrome” community. J. Community Genet. 2019, 11, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Dalglish Family 22q Clinic. Available online: http://22q.ca/ (accessed on 29 July 2020).
- The International 22q Foundation Inc. Available online: http://www.22q.org/ (accessed on 20 May 2020).
- Bassett, A.S.; McDonald-McGinn, D.; Devriendt, K.; Digilio, M.C.; Goldenberg, P.; Habel, A.; Marino, B.; Oskarsdottir, S.; Philip, N.; Sullivan, K.E.; et al. Practical Guidelines for Managing Patients with 22q11.2 Deletion Syndrome. J. Pediatr. 2011, 159, 332–339.e1. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.; Costain, G.; Ogura, L.; Silversides, C.K.; Chow, E.W.; Bassett, A.S. Reproductive Health Issues for Adults with a Common Genomic Disorder: 22q11.2 Deletion Syndrome. J. Genet. Couns. 2015, 24, 810–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didriksen, M.; Fejgin, K.; Nilsson, S.R.; Birknow, M.R.; Grayton, H.M.; Larsen, P.H.; Lauridsen, J.B.; Nielsen, V.; Celada, P.; Santana, N.; et al. Persistent gating deficit and increased sensitivity to NMDA receptor antagonism after puberty in a new mouse model of the human 22q11.2 microdeletion syndrome: A study in male mice. J. Psychiatry Neurosci. 2016, 42, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Beaton, E.A.; Simon, T.J. How might stress contribute to increased risk for schizophrenia in children with chromosome 22q11.2 deletion syndrome? J. Neurodev. Disord. 2010, 3, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Swillen, A.; Moss, E.; Duijff, S. Neurodevelopmental outcome in 22q11.2 deletion syndrome and management. Am. J. Med. Genet. Part A 2018, 176, 2160–2166. [Google Scholar] [CrossRef]
- Weaver, I.C.; Cervoni, N.; Champagne, F.A.; D’Alessio, A.C.; Sharma, S.; Seckl, J.R.; Dymov, S.; Szyf, M.; Meaney, M.J. Epigenetic Programming by Maternal Behavior. Nat. Neurosci. 2004, 7. [Google Scholar] [CrossRef]
- Szyf, M.; McGowan, P.; Meaney, M.J. The social environment and the epigenome. Environ. Mol. Mutagen. 2008, 49, 46–60. [Google Scholar] [CrossRef]
- Weisman, O.; Feldman, R.; Burg-Malki, M.; Keren, M.; Geva, R.; Diesendruck, G.; Gothelf, D. Mother–Child Interaction as a Window to a Unique Social Phenotype in 22q11.2 Deletion Syndrome and in Williams Syndrome. J. Autism Dev. Disord. 2015, 45, 2567–2577. [Google Scholar] [CrossRef]
- Shashi, V.; Keshavan, M.; Kaczorowski, J.; Schoch, K.; Lewandowski, K.E.; McConkie-Rosell, A.; Hooper, S.R.; Kwapil, T.R. Socioeconomic Status and Psychological Function in Children with Chromosome 22q11.2 Deletion Syndrome: Implications for Genetic Counseling. J. Genet. Couns. 2010, 19, 535–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olszewski, A.K.; Radoeva, P.D.; Fremont, W.; Kates, W.R.; Antshel, K.M. Is child intelligence associated with parent and sibling intelligence in individuals with developmental disorders? An investigation in youth with 22q11.2 deletion (velo-cardio-facial) syndrome. Res. Dev. Disabil. 2014, 35, 3582–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| miRNA: | Expression Level: | Tissue/Organ: | Possible Contribution to: |
|---|---|---|---|
| miR-185 | 0.4 times lower (H) lower (MM) different expression (H) *: CHD+/− hypocalcemia+/− low CD3 T cell count +/− | Synapses (H), hippocampus (MM) | Immune system deficiency, neurological abnormalities, hypotonia |
| miR-194 | lower (H, MM) different expression: CHD+/− hypocalcemia+/− low CD3 T cell count +/− | prefrontal cortex (MM), hippocampus (MM) | dendric and spine development deficits in hippocampus, acute myocardial infraction |
| miR-208 | higher(H) | no data | cardiac disease |
| miR-190 | higher(H) | no data | cardiac disease |
| miR-1 | higher (H) | no data | |
| miR-150 | lower (H) different expression: CHD+/− low CD3 T cell count +/− | no data | low number of mature T and B cells |
| miR-363 | lower (H) | no data | no data |
| miR-15b-3p, miR-324-5p, miR-720 | different expression: CHD+/− hypocalcemia+/− low CD3 T cell count +/− | no data | no data |
| miR-363, miR-23b | different expression: CHD+/− | no data | no data |
| miR-21 | different expression: hypocalcemia+/− low CD3 T cell count +/− | no data | no data |
| miR-145 | different expression: hypocalcemia+/− | no data | no data |
| miR-365-5p | different expression: CHD+/− low CD3 T cell count +/− | no data | no data |
| miR-29a | different expression: low CD3 T cell count +/− | no data | no data |
| Clinical Manifestation | Frequency in 22q11.2 DS | Human Phenotype Ontology Database * |
|---|---|---|
| Palatal anomalies | 69–100% | Cleft palate (HP:0000175), Abnormality of the pharynx (HP:0000600), Platybasia (HP:0002691) |
| Learning disabilities | >95% | |
| Speech delay | 79–84% | |
| Cardiac anomalies | 49–83% | Abnormality of cardiovascular system morphology (HP:0030680), Abnormal aortic arch morphology (HP:0012303), Truncus arteriosus (HP:0001660), Ventricular septal defect (HP:0001629), Abnormal pulmonary valve morphology (HP:0001641), Tetralogy of Fallot (HP:0001636), Atrial septal defect HP:0001631 |
| Immunodeficiency | 77% | Immunodeficiency (HP:0002721), Abnormality of the tonsils (HP:0100765), Hypocalcemia (HP:0002901), Impaired T cell function (HP:0005435) |
| Developmental delay in infancy | 75% | |
| Ophthalmologic abnormalities | 7–70% | Posterior embryotoxon (HP:0000627), Corneal neovascularization (HP:0011496), Ptosis (HP:0000508) |
| Endocrine | 60% | Hypoplasia of the thymus (HP:0000778), Hypoparathyroidism (HP:0000829) |
| Behaviour/psychiatric problems | 9–50% | |
| Developmental delay in childhood | 45% | Short stature (HP:0004322) |
| Renal anomalies | 36–37% | Renal hypoplasia (HP:0000089) |
| Feeding and Swallowing Problems | 35% | Anorectal anomaly (HP:0012732), Constipation (HP:0002019) |
| Skeletal abnormalities | 17–19% | Arachnodactyly (HP:0001166), Abnormal skull morphology (HP:0000929), Short neck (HP:0000470) |
| Neurologic | 8% | |
| Dental: Delayed eruption, Enamel hypoplasia | 2.5% | Abnormality of the dentition (HP:0000164), Carious teeth (HP:0000670) |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karbarz, M. Consequences of 22q11.2 Microdeletion on the Genome, Individual and Population Levels. Genes 2020, 11, 977. https://doi.org/10.3390/genes11090977
Karbarz M. Consequences of 22q11.2 Microdeletion on the Genome, Individual and Population Levels. Genes. 2020; 11(9):977. https://doi.org/10.3390/genes11090977
Chicago/Turabian StyleKarbarz, Małgorzata. 2020. "Consequences of 22q11.2 Microdeletion on the Genome, Individual and Population Levels" Genes 11, no. 9: 977. https://doi.org/10.3390/genes11090977
APA StyleKarbarz, M. (2020). Consequences of 22q11.2 Microdeletion on the Genome, Individual and Population Levels. Genes, 11(9), 977. https://doi.org/10.3390/genes11090977