1. Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative neoplasm caused by reciprocal translocation t(9;22)(q34;q11), resulting in the formation of the Philadelphia chromosome and
BCR-ABL1 fusion oncogene [
1,
2]. The hybrid gene
BCR-ABL1 undergoes translation into chimeric protein, which is a constitutively active tyrosine kinase which phosphorylates several target proteins and in effect enables expansion of leukemic stem and progenitor cells. Natural course of the disease progression is characterized by a successive increase in the number of blast cells in the blood and bone marrow and is classified into phases: chronic phase (CP-CML), accelerated phase (AP-CML), and blastic phase (BP-CML), also called blast crisis. Although the introduction of tyrosine kinase inhibitors (TKIs) to the therapy of CML significantly improved the outcome for the great majority of patients, there is still a minor group of patients who develop drug resistance and are at risk of progression. The pathogenesis of BP-CML is still poorly understood and TKIs have limited effectiveness in this phase of the disease [
3,
4]. One of the features of BP-CML is genomic instability when leukemic stem cells acquire additional genetic changes that may cause drug resistance and lead to disease relapse [
5].
Telomere maintenance is crucial for the genomic stability of normal cells, and among several possible mechanisms leading to genomic instability in cancer cells, disrupted telomere maintenance is one of the hallmarks [
6]. Telomeres (in eukaryotes termini of chromosomes) are composed of tandem repeats of six base pairs (TTAGGG) and, together with several proteins, named shelterin complex, protect chromosome ends from recognition by DNA repair machinery as double strand breaks (DSBs) and from end to end fusion [
7]. In human cancer, telomere shortening and aberrant activation of telomerase is one of the key features of oncogenic transformation [
8]. Telomere length is regulated by telomerase complex, which consists of telomerase reverse transcriptase (TERT) and two copies of RNA template (TERC) and also additional proteins stabilizing the complex, such as dyskerin (DKC1) and others (NHP2, NOP10 and GAR1). Telomere maintenance in malignant cells is regulated not only by expression of telomerase complex, but also by various telomere-associated proteins, such as the shelterin complex [
9]. The major role of shelterins is to prevent the recognition of telomeres as DNA damage sites. The complex is composed of six proteins: telomeric repeat-binding factors 1 and 2 (TRF1 and TRF2), TRF1-interacting nuclear factor 2 (TIN2), protection of telomeres (POT1), POT1 and TIN2-interacting protein 1 (TPP1), and TRF2-interacting protein 1 (RAP1) [
9]. Additionally, other telomeric-associated proteins, such as TEP1 and tankyrase, interact with the shelterin complex. In general, TERT complex and tankyrase are considered as positive regulators of telomere length, while TRF1, TRF2, and POT1 are negative regulators [
9].
In CML, telomere attrition has been associated with disease progression [
10]. Telomeres are significantly shorter in BP-CML patients’ cells as compared to cells from CP-CML, while the latter are shorter than in cells from healthy donors [
11,
12]. It has been proposed that telomere shortening can be considered as a prognostic marker in CML [
13]. Interestingly, in contrast to many advanced solid tumors, TERT expression is rather downregulated in BP-CML as compared to CP-CML and reduced TERT expression has been attributed to telomere shortening in CML patients [
14]. Therefore, other mechanisms than the activation of TERT are possibly involved in dealing with critically short telomeres, especially in BP-CML.
The aim of this study was to investigate expression and activity of genes involved in different mechanisms of telomere maintenance as well as mutational status of the most significant members of the telomerase complex and shelterins, in widely used CML cell lines. Additionally, a possible link between aberrant telomere regulation and genomic instability in BP-CML cells has been examined. We employed five well-established BCR-ABL1-positive cell lines, all derived from BP-CML patients, namely K562, KU-812, LAMA-84, MEG-A2, and MOLM-1, and additionally HL-60 as BCR-ABL1-negative acute myeloid leukemia cell line as a control.
2. Materials and Methods
2.1. Cell Lines
All human cell lines (HL-60, K562, KU-812, LAMA-84, MEG-A2 and MOLM-1) were obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Braunschweig, Germany). HL-60, K562, LAMA-84 were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS). KU-812, MOLM-1 were cultured in RPMI-1640 medium supplemented with 20% FBS and MEG-A2 were cultured in IMDM medium supplemented with 20% FBS. All media were supplemented with antibiotic solution (100 U/mL penicillin, 0.1 mg/mL streptomycin and 0.25 µg/mL amphotericin B) (all from Life Technologies, Carlsbad, CA, USA). Cells were maintained at 37 °C in 5% CO2 in a humidified incubator and were routinely tested for mycoplasma contamination using a PCR-based test.
2.2. Telomere Length Analysis and Telomerase Activity
Telomere length was analysed by two different methods, Southern blotting analysis of the genomic DNA and additionally at the level of the single cell by fluorescence in situ hybridization (FISH). The genomic DNA was extracted using the Wizard
® Genomic DNA Purification Kit (Promega, Madison, WI, USA) according to the protocol provided by the manufacturer’s recommendations. Average telomere length was measured by Southern blot analysis of terminal restriction fragments (TRF) using the TeloTAGGG telomere length assay kit (Roche, Basel, Switzerland) according to the manufacturer’s instructions as described previously [
15]. Mean TRF length for each cell line was estimated on the base of the highest signal intensity peak from TRF due to multiple hybridization of telomeric-specific hybridization probe. Densitometric profile was performed to correspond to bands of DNA marker using ImageJ with gel analysis module. Metaphase spreads and interphase nuclei were obtained from routine 24 h cell cultures according to protocol described in 2.1 material and methods section. To stop cell division the colchicine was added to a final concentration of 0.02 μg/mL and then incubate at 37 °C for a 2 h. After incubation, cells were treated with 75 mM KCl at 37 °C for 15 min and fixed in ethanol and glacial acetic acid (3:1) solution. Hybridization was performed with the Telomere PNA FISH kit/Cy3 (Dako, Glostrup, Denmark) according to the protocol provided by the manufacturer’s recommendation. Fluorescent signals were visualized under the Olympus BX61 with objective 40× and at least 100 interphase nuclei were analysed to determine the telomere lengths in each cell line. Telomere intensities were analysed using TFL-TELO Software, version 2.0, software (British Columbia Cancer Center, Vancouver, Canada). Telomerase activity was measured with a TeloTAGGG Telomerase PCR ELISA kit (Roche, Basel, Switzerland) according to the manufacturer’s instructions.
2.3. Fluorescence In Situ Hybridization (FISH)
FISH was performed using probes for: BCR-ABL1 t(9;22) Fusion (KBI-10005, Kreatech, Amsterdam, The Netherlands), JAK2 (9p24) Break (KBI-10012, Kreatech, Amsterdam, Netherlands), PDGFRB (5q32) Break (KBI-10004, Kreatech, Amsterdam, The Netherlands), TERT (5p15) (KBI-40113, Kreatech, Amsterdam, The Netherlands) or TERC (3q26)/3q11 (KBI-10110, Kreatech, Amsterdam, The Netherlands). For FISH experiments, a standard protocol was used. In brief, the cells were fixed with ethanol and glacial acetic acid (3:1) solution and treated with RNAse (100 µg/mL) in 2 × SSC buffer for 1 h at 37 °C in humidity chamber. After washing, first in PBS and then in PBS with 50 mM MgCl2, the slides were dehydrated in ethanol series. FISH probe was denatured together with the slide at 80 °C for 7 min and hybridized overnight in the dark at 37 °C in humidity chamber. The slides were washed, first at 72 °C and then at RT (0.4% Igepal in 2 × SSC and 2% Igepal in 2 × SSC, respectively). For DNA visualization, the slides were counterstained with a drop of mounting medium with 4′,6′-diamino-2-phenylindole counterstain (ProLong™ Diamond Antifade Mountant with DAPI, Invitrogen™ (Carlsbad, CA, USA). Fluorescent signals were visualized under the Olympus BX61 and MetaSystem Isis software (Altlußheim, Germany) with objective 40×. 100 interphase cells were examined. The signal pattern was analysed according to the manufacturer’s instructions.
2.4. Western Blotting
Cell pellets were lysed with ice-cold RIPA buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 1 mM PMSF and 1× complete protease inhibitors, Roche, Basel, Switzerland) and were boiled in 2 × Laemmli’s sample buffer. Lysates were separated by 10% SDS-PAGE at 120 volts for 60 min (Bio-Rad, Hercules, CA, USA) and transfer by electroblotting (Bio-Rad, Hercules, CA, USA) onto polivinylidene difluoride (PVDF) membrane (Merck Millipore, Burlington, MA, USA) at 4 °C at a constant voltage of 100 V for 100 min in a Towbin buffer (25 mM Tris, 192 mM glycine, 0.1% SDS and 20% ethanol). Membrane was blocked in TBS-T (Tris-buffered saline-Tween20) buffer containing 2% BSA for 60 min at room temperature and then incubated overnight at 4 °C in 1% BSA in TBS-T containing primary antibody: anti-phospho-CRKL (Tyr207) (1:500, #3181), anti-CRKL (1:500, #3182), anti-POT1 (1:1000, MAB5299), anti-RAP1 (1:1000, PA5-35137), anti-TERT (1:1000,ab32020), anti-TRF1 (1:1000, NB100-1701), anti-TRF2 (1:1000, P5-19426,), anti-HSP70 (1:1000, PA5-14521), anti-HSP90 (1:1000, MA1-10373) or anti-β-Actin (1:40,000, A3854). After three washes for 5 min each in TBS-T, they were incubated for 60 min at room temperature with secondary antibody (#7076S or #7074S) diluted 1:2000 in 1% BSA in TBS-T, and washed as described above. Antibodies were acquired from Cell Signaling Technology (Danvers, MA, USA), R&D Systems (Minneapolis, MN, USA), Abcam (Cambridge, United Kingdom), Novus Biologicals (Centennial, CO, USA) and Invitrogen™ (Carlsbad, CA, USA). The chemiluminescence signals were detected with an ECL Prime Western blotting Detection Reagent (GE Healthcare, Chicago, IL, USA) and a G:BOX imaging system (Syngene, Bangalore, India). Densitometry analysis was conducted using ImageJ software (
https://imagej.nih.gov/ij/) (U. S. National Institutes of Health, Bethesda, MD, USA).
2.5. Immunofluorescence
For promyelocytic leukemia protein (PML) and TRF2 co-localization, interphase nuclei were used. The cells were fixed with 3.7% formaldehyde in PBS for 20 min in 1.5 mL tube. After 3 times washing with PBS, the cells were transferred on Polysine Slides (Thermo Scientific™ Shandon™ Polysine Slides, Thermo Scientific, Waltham, MA, USA) and permeabilized with 0.3% Triton X-100 in PBS for 5 min at RT and next blocked with 1% BSA in PBS-T (PBS-Tween20) at RT for 30 min. After washing with PBS-T, the cells were incubated with antibodies: anti-PML (1:200, ab96051) and anti-TRF2 (1:100, ab108997) (Abcam, Cambridge, UK) overnight at 4 °C. After incubation, cells were washed twice with PBS and incubated with secondary antibodies: FITC-conjugated and TexasRed-conjugated, respectively (all at 1:000, F2761, T6390) (Life Technologies, Carlsbad, CA, USA) for 1 h at RT in the dark. Cells were then washed three times with PBS and nuclei were stained with DAPI. Images were taken using an Olympus BX61 fluorescent microscope (Shinjuku, Japan) with objective 20×. To analyze co-localization PML/TRF2, ImageJ software
http://rsbweb.nih.gov/ij/ with JACoP plugin was used [
16]. The Pearson’s coefficient was used to calculating a set of co-localization indicators.
2.6. DNA Damage and DNA Damage Response
DNA double strand breaks (DSBs) were analyzed using neutral comet assay upon stimulation with DSBs inducer: 10 µM etoposide for 24 h, as described previously [
17]. The cells were cultured according to the protocol described in the Material and Methods section. The all cell cultures including positive control with etoposide treatment as well as negative control without etoposide were performed in three independent experiments after 24 h cell culture. The percentage of tail DNA was used as a parameter of DNA damage. Images were taken using an Olympus BX61 fluorescent microscope with objective 20×. Comets (
n = 100) were analyzed using CASP, an open-source software tool 1.2.3beta2 version (
http://casplab.com) (Wroclaw, Poland). The activation of ATM and H2AX was measured using flow cytometry and Muse™ Multi-Color DNA Damage kit (Merck Millipore, Burlington, MA, USA) as described elsewhere [
18].
2.7. Detection of Micronuclei (MN)
The slides were fixed with ethanol and glacial acetic acid (3:1) solution and were stained with DAPI. The results were expressed as the number of MN-positive cells per 100 nuclei (%).
2.8. Real-Time PCR
The reverse transcription quantitative PCR (RT-qPCR) was used to evaluate expression level of the following genes: BCR-ABL1, TRF1, TRF2, TERT, TERC, RAP1 and POT1. RNA extraction (TriPure Isolation Reagent, Sigma-Aldrich, Saint Louis, MO, USA) followed by reverse transcription (Transcriptor First Strand cDNA Synthesis Kit, Roche, Basel, Switzerland) were performed according to the manufacturer’s manuals. RT-qPCR was carried out in LightCycler™ 480 instrument (Roche, Basel, Switzerland) using LightCycler® 480 Probes Master and Universal Probe Library (Roche, Basel, Switzerland) in a final volume of 10 µL. β2M and GUSB were used as reference genes.
2.9. Next-Generation/Sanger Sequencing
Mutational analysis of coding sequences of genes associated with haematological malignancies as well as typical tumor suppressors and oncogenes was carried out using targeted enrichment by one of our two custom-made gene panels (targeting genes relevant to human malignancies). Lists of sequenced genes are presented in
Supplementary Table S1. Genomic DNA was isolated using Gentra Puregene Cell Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. DNA libraries were prepared from 0.5 μg DNA using Kapa Library Preparation Kit for Illumina (Roche, Basel, Switzerland), multiplexed with others before solution-based capture (Roche NimbleGen, Basel, Switzerland). The capture protocol was performed according to the manufacturer’s recommendations. Briefly, 1 μg of pooled DNA was hybridized for 72 h with the probes, then after washing steps, the captured DNA was amplified, purified, qualified using Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and quantified using Qubit (Life Technologies, Carlsbad, CA, USA). Cell lines were sequenced on Illumina HiSeq 1500 (Illumina, San Diego, CA, USA) to achieve mean depth of coverage in range 63.9–107.6× followed by GATK 4.0.1.1-based variant discovery [
19]. Variant discovery comprised of quality control of raw fastq, adapter trimming and low quality reads removal using Trimmomatic [
20], read mapping to hg19 genome using BWA-MEM 0.7.15-r1140 [
21], duplication removal, local realignment and quality recalibration using GATK and Picard and variant calling using HaplotypeCaller. Variants more common than 1% in public (1000 genomes, NHLBI ESP, gnomAD) databases as well as variants more common than 5% in our internal database were removed from the analysis. To identify functional consequences of detected variants five bioinformatics predictors were used: CADD, PolyPhen2, SIFT, FATHMM and Mutation Taster. Copy-number calling was performed by sequencing coverage analysis using CNVkit v0.9.5 [
22]. Non-leukemic blood samples and leukemic blood samples verified as devoid of detectable copy number variations were used to create coverage references for panels 1 and 2, respectively. CNVkit was run with default settings, except that bin size was limited to 400 bp to increase resolution for both panels. Additionally, off-target coverage was not used in CNV calling for panel 2. CNVkit built-in function center-at was used to correct log2 coverage ratio values in samples with copy-number-neutral regions incorrectly placed under 0 (for KU-812 and MEG-A2,
Supplementary Figures S8 and S9,
Supplementary Table S2).
TERT promoter hotspot mutations (C228T and C250T) were analysed by NGS, but due to low coverage of the promotor region, also additionally confirmed by Sanger sequencing as described [
23], with modifications. Briefly, primer sequences were as follows: TERT-F: AGGAGGCTCTTTGGAGCAAG, TERT-R: CTCTGAACTCTGTGCTGACCA. PCR was performed using KAPA HiFi HotStart ReadyMix PCR Kit (Roche, Basel, Switzerland) with the program 3 min at 95 °C followed by 35 cycles of 20 s at 98 °C, 10 s at 71 °C and 10 s at 72 °C and final extension at 72 °C for 5 min. Before sequencing on a ABI3730 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA), the PCR products were purified using Agencourt AMPure XP beads (Beckman Coulter, Brea, CA, USA) and labelled with BigDye Terminator v.3.1 (Applied Biosystems, Foster City, CA, USA).
2.10. Statistical Analysis
Statistical significance was assessed using GraphPad Prism 6.07 (GraphPad Software, Inc., La Jolla, CA, USA) by one-way ANOVA and with the Tukey’s multiple comparison test. Differences between control conditions versus etoposide treatment were assessed with the one-way ANOVA with the Dunnett’s multiple comparison test. The correlation analysis was performed using linear correlation (Pearson r) test. p-Values of less than 0.05 were considered significant.
4. Discussion
According to the current consensus, average telomere length in leukemic cells of CML patients is shorter as compared to white blood cells of age-matched healthy individuals [
10,
11,
12]. Furthermore, telomere shortening was linked to disease progression from chronic to blastic phase of CML, with a shortening rate approximately 10 times higher than in normal controls [
13]. Patients with a high-risk Hasford score at diagnosis exhibited significantly greater telomere loss than patients with a low-risk score, while patients with intermediate risk showed an intermediate telomere loss rate [
13]. Telomere shortening is more pronounced in leukemic stem cells as compared to normal hematopoietic stem cell compartment at diagnosis as described by Bouillon et al. [
30]. It has also been shown that accelerated shortening of telomeres in CML cells can be accompanied by the “telomere-associated secretory phenotype”, driven by RAP1, but not by BCR-ABL1 kinase, however mechanistic explanation of this phenomenon is not complete [
31]. Other studies showed that chromosome ends may be unchanged or even elongated in some CML patients. Longer telomeres of chromosome arms have been reported in CML patients compared with healthy donors. Long telomeres may contribute to the cell proliferation during clonal selection in the early stage of CML ontogenesis [
13]. This phenomenon can be explained by the mechanisms of alternative lengthening of telomeres (ALT). It has been proposed that ALT plays an important role in early phases of CP-CML, but not in BP-CML [
32]. Since it has been postulated that telomerase activity increases with CML progression, the dominating telomere maintenance mechanism might undergo transition from ALT to telomerase-dependent. Therefore, both the fate of the telomeres and the origin of telomere shortening in CML cells are not fully understood.
In the current work, we employed five
BCR-ABL1-positive cell lines most commonly used in the laboratories for the studies on CML molecular pathogenesis and to test new therapeutic approaches. As a control, we used HL-60 cell line, a
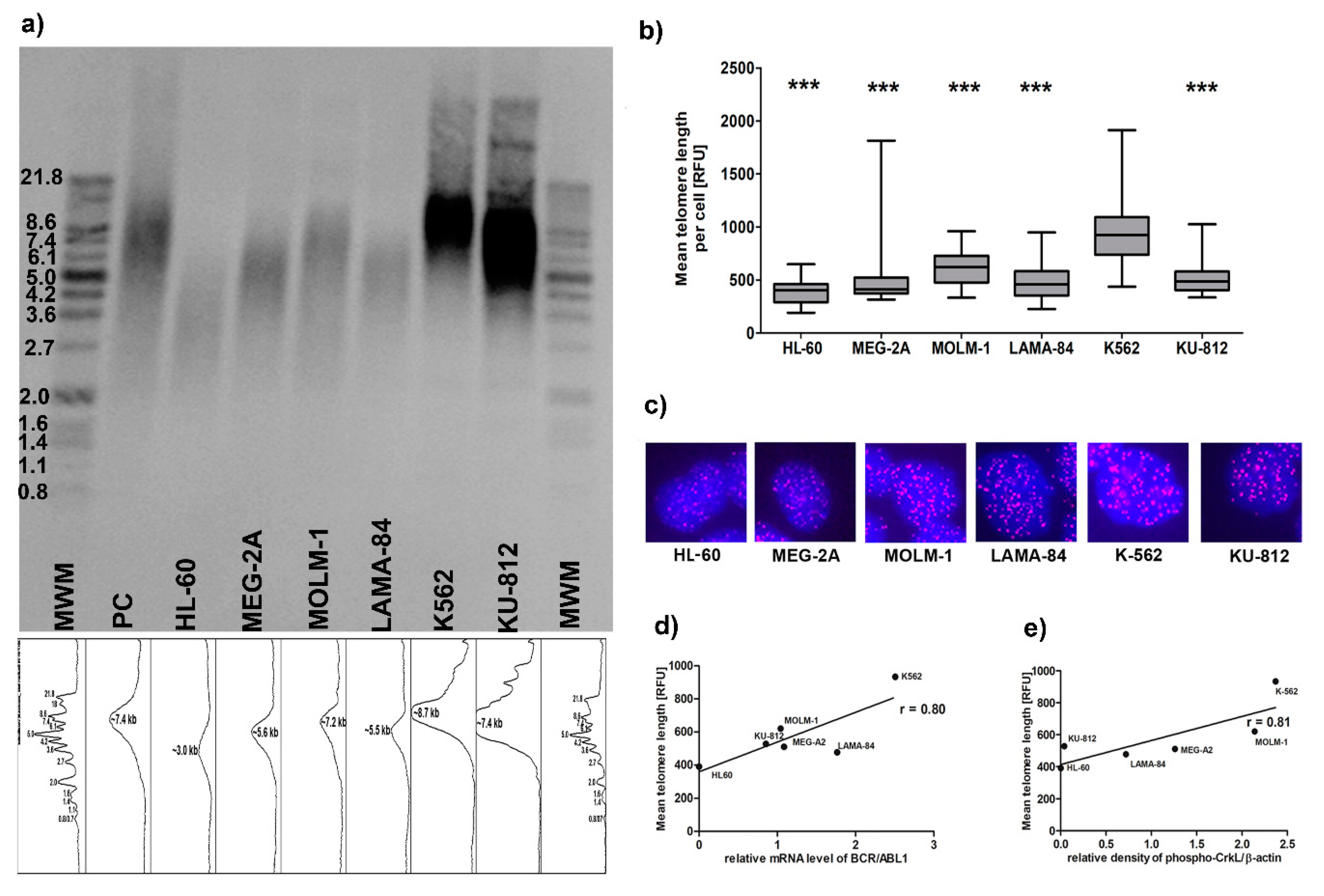
BCR-ABL1-negative acute leukemia, since normal healthy CD34+ bone marrow cells are not considered as a well-suited control cells for BP-CML cell lines harboring several genetical abnormalities. Our results show that telomeres in cell lines derived from acute phases of CML have different lengths, but all are significantly longer than
BCR-ABL1-negative HL-60 cells. However, between the CML cell lines telomere length positively correlates with both
BCR-ABL1 expression and activity. K562, which had longest telomeres, demonstrated high level of amplification of
BCR-ABL1 detected at the single cell level by FISH and confirmed by CNV analysis form NGS data and also had highest activity of BCR-ABL1 kinase (
Figure 1 and
Figure 2 and
Supplementary Table S2). FISH analysis was applied to other genes known to be dysregulated in myeloid malignancies such as
JAK2 and
PDGFRB.
JAK2 gene is a known activator of telomerase and JAK2 inhibitors reduced the telomerase activity [
33]. The study of
PDGFRB gene is of importance because TRF2 regulates expression of
PDGFRB gene by binding and activating the
PDGFRB promoter as an example an extra-telomeric function of TRF2 [
24]. Although we have found that the copy number of
JAK2 differed between cell lines, it seems irrelevant to telomere maintenance. Similarly, the higher number of
PDGFRB gene copies also was not associated with longer telomeres or with higher expression of TRF2. The major enzyme required for the addition of telomeric repeats to the termini of eukaryotic chromosomes is telomerase. Human telomerase consists of a human telomerase RNA component (
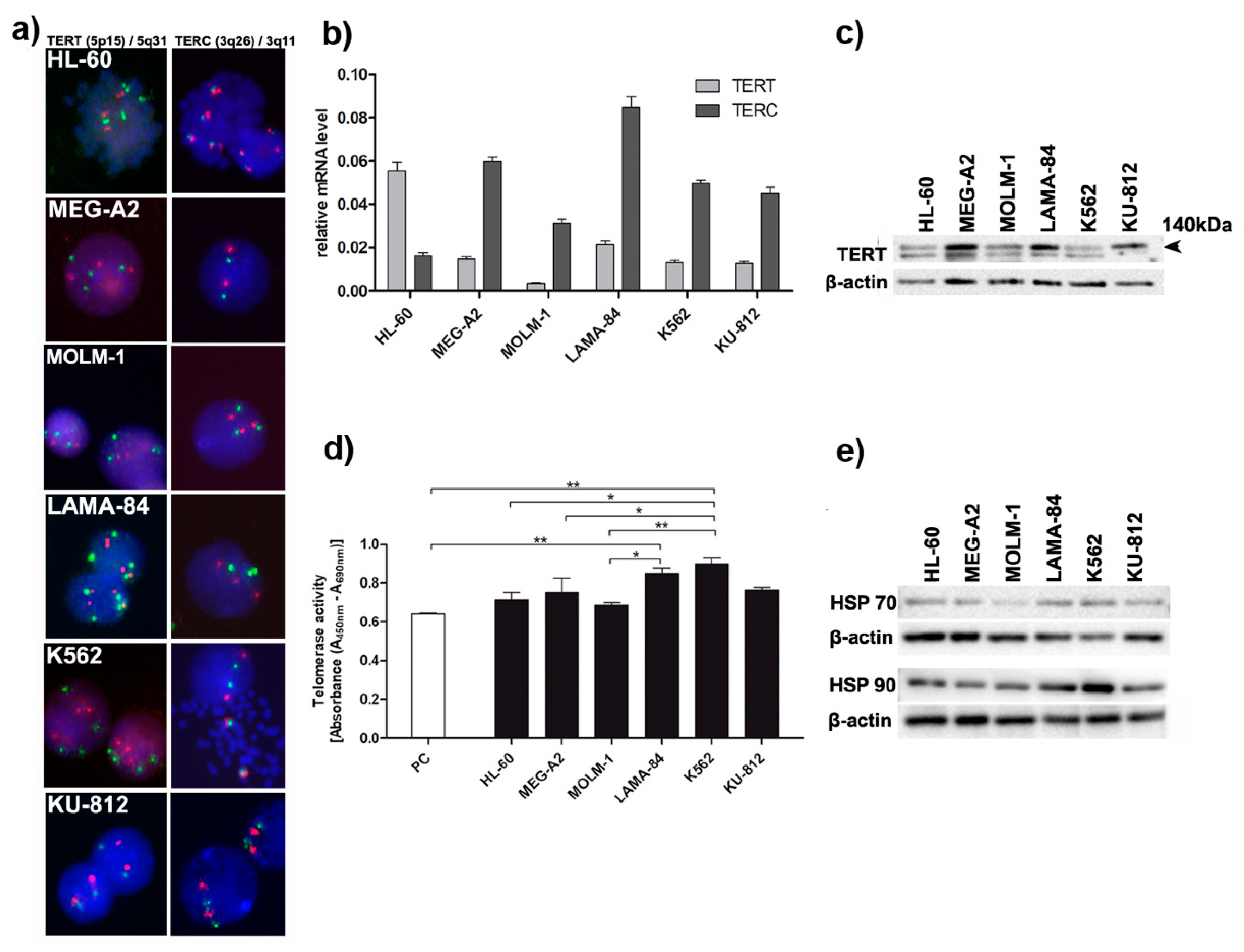
TERC), a human telomerase catalytic subunit (TERT) and TEP1, a telomerase associated protein. Previous studies showed that, in CML cells, the expression of
TERT may be downregulated [
14]. Our results showed that although the expression levels of
TERT and
TERC gene were generally low in all studied cell lines, K562 cells again demonstrated highest enzymatic activity of telomerase, as compared to other cell lines and highest protein level of both chaperones HSP70 and HSP90 necessary for telomerase activity (
Figure 3).
We have already characterized the global genomic landscape of K562 cell line employing targeted enrichment and next-generation sequencing [
27]. In this work, we detected several pathogenic variants, which are described already (such as in
TP53), but also several new pathogenic variants not previously described in DNA repair genes (such as
BRCA1 or
MLH1). This may contribute to the aggressive phenotype and genomic instability of this cell line. In the current work, we applied FISH analysis at the level of the single cell and targeted sequencing of several genes involved in human cancer by NGS for the whole panel of cell lines to characterize complex genetic aberrations underlying observed differences in telomere maintenance and telomeric complex genes (
Table 1). We did not find any significant defects in the relevant genes involved in telomere maintenance except one pathogenic variant in
TERT gene in K562 cells (
Table 1,
Supplementary Figure S3), which we already published [
27]. However, we observed inactivation of the major tumor suppressors function, including
TP53 gene (by chromosomal deletion or combination of pathogenic mutation and second allele loss) in all studied cell lines as well as in
CDKNA2 gene encoding p16
INK4a. The analysis of genetic status of
TP53 and
CDKN2A is crucial for cellular telomere-damage response pathway in many cells. It is well known that p16
INK4a contributes to the p53-independent response to telomere damage and depends from genetic status of p16
INK4a and p53, thus, two distinct pathways could be activated [
34]. Moreover, it can be postulated that the elongation of telomeres is associated with the loss of p53 and p16
INK4a function [
35]. This may explain how the cells escape from telomere-mediated cell senescence and/or apoptosis in the BP-CML cell lines, since inactivation of
TP53 was already implicated as a responsible mechanism [
36].
Analysis of the expression of selected genes, crucial components the telomeric complex, such as shelterins, revealed that BCR-ABL1 kinase expression and activity may upregulate the level of RAP1 and POT1, as well as TRF1, as demonstrated by positive correlations (
Figure 4). This is partially in line with data published by Braig et al. pointing at the important role of RAP1 in telomere maintenance in CML cells [
31].
Our results demonstrated that the differences in telomere length between
BCR-ABL1-positive cell lines can be explained only partially by telomerase activity (
Figure 3). Another molecular mechanism, which may be responsible for observed variation in telomere lengthening is the alternative lengthening of telomere (ALT) phenomenon. The role of shelterin proteins in alternative lengthening of telomeres in chronic myeloid leukemia has not been studied in detail up to now, therefore we have decided to analyze co-localization of alternative lengthening of telomeres-associated promyelocytic leukemia (PML) nuclear bodies (APBs) with TRF2 commonly used as a marker of ALT. The APB contains telomeric DNA, extrachromosomal telomeric circles (t-circles), DNA repair proteins and the telomere repeat binding factors such as TRF2 [
37,
38,
39]. Our immunofluorescence analysis showed that TRF2 was co-localized with PML only in K-562 cells (
Figure 5). These results may suggest that telomere length in K562 despite activity of telomerase may be also associated with ALT phenomenon, which is confirmed by previous study showing that the t-circles, one of the ALT hallmarks, can be used to define ALT activation in CML patients [
11]. Samassekou et al. have shown that in 27% CML-CP cells exhibited both high ALT activity and telomerase activities. As telomerase activity increases with disease progression, the dominating telomere maintenance mechanism might undergo transition from ALT to telomerase. In this context, both the consequence and the origin of telomere shortening are still a matter of debate.
Telomere maintenance dysfunction in cancer cells is commonly associated with genomic instability [
40]. We have used common markers to monitor the genomic instability, such as micronuclei, amount of DNA in Tail (comet assay), or recruitment of H2AX (
Figure 5). In this study, we tested the hypothesis that upregulation of telomeric proteins may result in increased pool of telomere-unbound proteins as an adaptation to oxidative stress conditions. This may promote the inhibition of ATM/ATR and cause the attenuation of DNA damage response (DDR), e.g., limited recruitment of H2AX, which may lead to an increase in the global genomic instability and clonal selection (
Figure 5). Surprisingly, we have found that level of micronuclei production (
Figure 1) and number of double strand breaks as measured by comet assay (
Figure 5) in cells without exogenic stress were not associated with
BCR-ABL1 copy number. However, upon etoposide treatment, the activation of ATM kinase in K562, LAMA-84 and MEG-2A cell lines was more pronounced than in HL-60 cells. Simultaneously the histone H2AX phosphorylation on Ser139 in response to DNA damage was less pronounced in K562 than in KU-812 and MOLM-1 (
Figure 5).
This suggests that BCR-ABL1 translocates to the nucleus and associates with ATM after DNA damage but does not affect ATM function. This above observation is agreed with previous report showing that BCR-ABL1 disrupts ATR-dependent signal transduction events but not ATM function [
41]. Additionally, we have noted that the lower phosphorylation of the histone H2AX on Ser139 in response to DNA damage corresponded with higher TRF2 level in K562 (
Figure 4 and
Figure 5). This could be result of blunting of the DNA damage response in K562 cells when TRF2 is overexpressed [
42]. Interestingly, the comparison analysis of the levels of RAP1 identified as a TRF2-interacting protein between cell line has not revealed any significant relationship with the comet DBS assay, the micronucleus counts, phosphorylation of the histone H2AX on Ser139 in response to DNA damage (
Figure 1,
Figure 4, and
Figure 5). The LAMA-84 cell line that has the one of higher level of RAP1 expression has almost similar level of DNA damage (micronuclei and DSB) compared to KU-812 or MEG-A2, cell lines with the lowest level of RAP1. Similar observation was noted upon etoposide treatment (
Figure 5). Moreover, it is noted that K562, the cell line with the highest level of RAP1 and other shelterin proteins, has not shown any significant changes in the levels of genomic instability compared to other CML cell lines upon etoposide treatment (
Figure 5). So, our results have not confirmed previous observation e.g., in gastric cancer cells treated with etoposide and we cannot conclude that shelterin proteins play important role in etoposide-mediated DNA damage response in CML cells [
43]. The correlational analysis between telomere length and RAP1 or TFR2 level in analyzed cell line has revealed that is not significant correlation between telomere length with RAP1 (r = 0.68) and TRF2 level (r = 0.47) respectively. On the other hand, RAP1 is a well-known factor of DNA damage response, not only in telomeric, but also non-telomeric regions, so it is possible that RAP1-mediated genome stability may be based on its other protective role not related to direct telomere function in CML cell lines [
44]. It was shown that mammalian Rap1 could be important for hematopoietic stem cell survival or response to chemotherapy through e.g., interaction between XRCC4/DNA Ligase IV and DNA-PK [
45]. So, we conclude that only losing of RAP1 or other shelterin proteins like TRF1 or TRF2 could decrease double-strand break repair, but overexpression of these proteins is not able to promote clastogenic effects. Similar effect we have already observed in human dermal fibroblasts, HeLa, ACHN, A549, and MCF7 cells as we have also detected the overexpression of TRF1 and TRF2 as a result of stress condition. Simultaneously, we have observed limited recruitment of 53BP1 without any telomere length changes [
46]. Therefore, we supposed that upregulation of TRF proteins results in an increased pool of telomere unbound TRFs that regulate the expression of subtelomeric genes as part of cellular adaptive response, however, this phenomenon should be studied in detail in the future.
Blastic phase of CML, though rare in the era of TKI, remains incurable except patients in whom allogeneic hematopoietic stem cell transplantation was successful. Studies on molecular pathogenesis of BP-CML showed high level of genomic instability and additional genetic aberrations, independent of BCR-ABL1 activity [
3] also in leukemic stem cells compartment [
5,
47]. While this knowledge helped in our understanding of the molecular mechanisms of drug resistance and/or progression, it did not translate yet into effective targeting and treatment of patients in acute phase of CML. Unraveling the mechanisms of telomere maintenance and the role of telomerase and shelterin complex in BCR-ABL1-mediated genomic instability may contribute to the development of new strategies preventing or counteracting resistance phenotype and malignant progression of the disease. Targeting telomerase in acute myeloid leukemia proved that strategy aiming at the aberrant regulation of telomere maintenance may help to eradicate leukemic stem cells [
48].
In summary, this work focused on BP-CML cell lines shows a complex picture of the regulation of telomere length and expression of telomere-related genes in BP-CML cells supporting the crucial role of BCR-ABL1 kinase expression and activity in the maintenance of telomeres. Presented results may help to validate and properly interpret results obtained by many laboratories employing these widely used cellular in vitro models of CML. Additionally, our data can be applied in the search for new therapeutic opportunities not only for advanced CML, but also in other acute leukemias.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}