Merging Genomics and Transcriptomics for Predicting Fusarium Head Blight Resistance in Wheat

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Statistical Analysis of the Phenotypic Data
2.3. Genotypic Data and Transcriptome Profiling
2.4. Single-Trait Omics-Based Prediction
2.5. Trait-Assisted and Single-Step Prediction Models
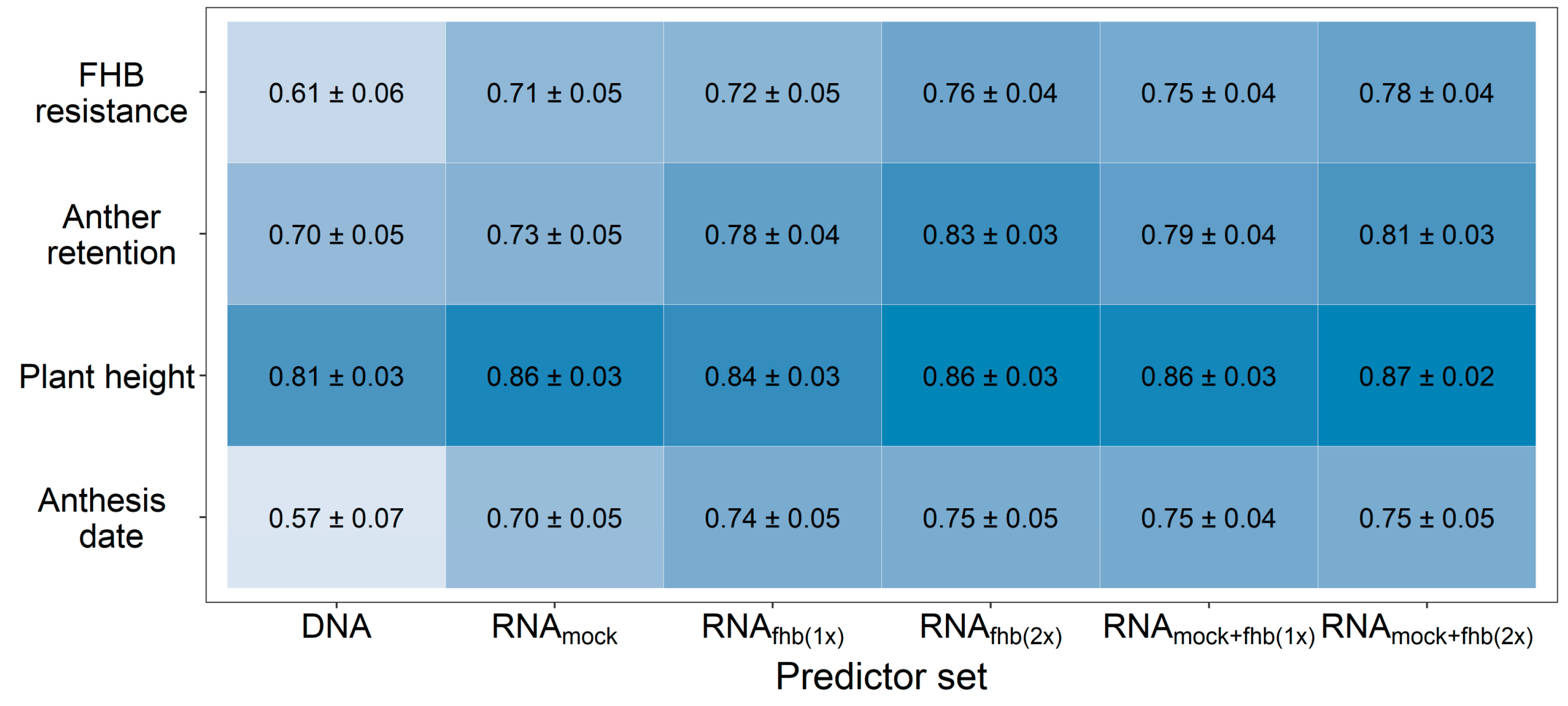
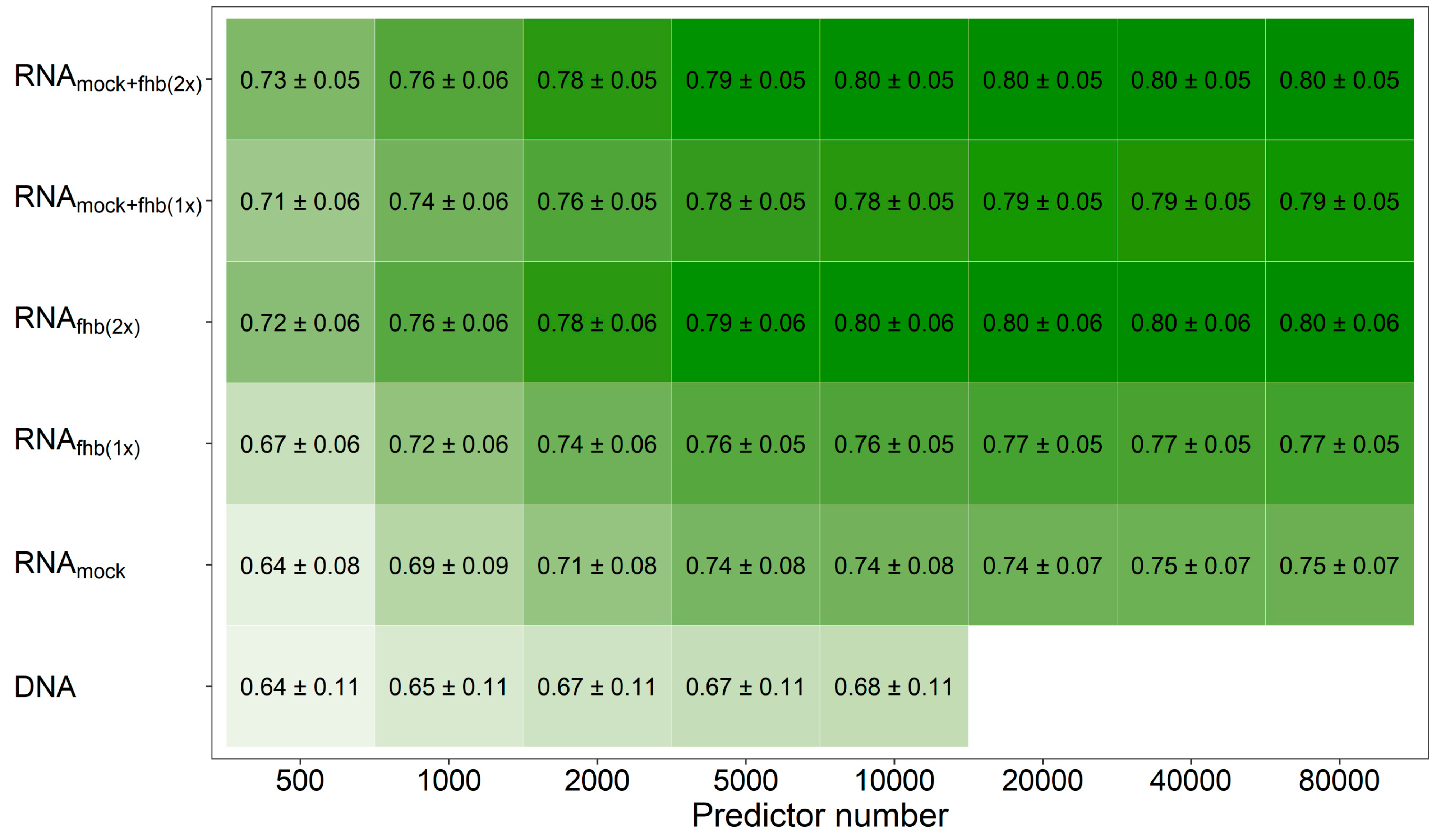
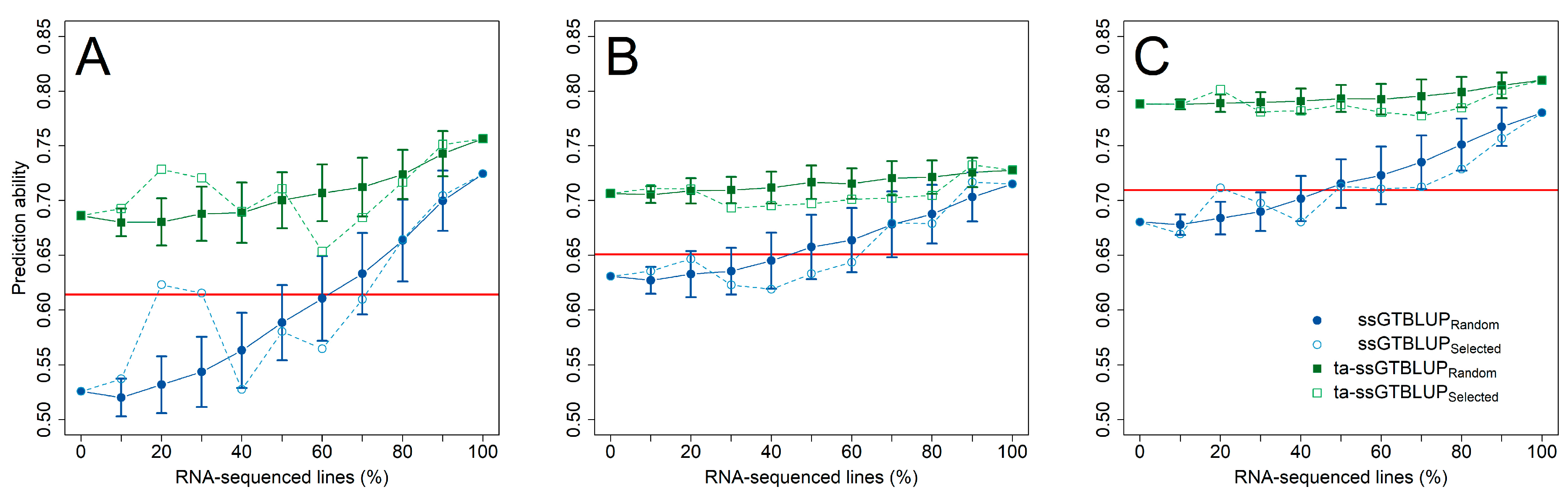
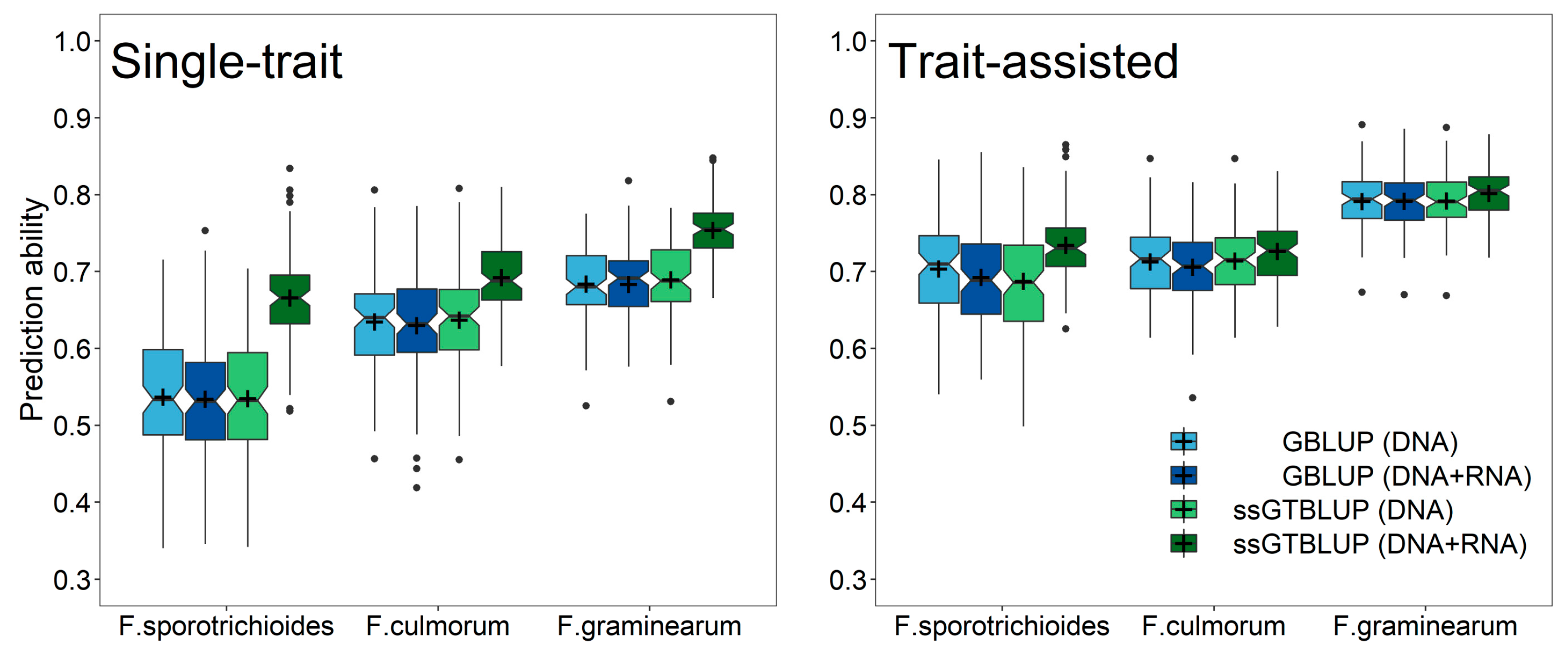
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Robertsen, C.; Hjortshøj, R.; Janss, L. Genomic Selection in Cereal Breeding. Agronomy 2019, 9, 95. [Google Scholar] [CrossRef] [Green Version]
- Poland, J.; Rutkoski, J. Advances and Challenges in Genomic Selection for Disease Resistance. Annu. Rev. Phytopathol. 2016, 54, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Haile, J.K.; Diaye, A.N.; Sari, E.; Walkowiak, S. Potential of Genomic Selection and Integrating “Omics” Data for Disease Evaluation in Wheat. Crop. Breed. Genet. Genom. 2020, 2, e200016. [Google Scholar] [CrossRef]
- Ma, Z.; Xie, Q.; Li, G.; Jia, H.; Zhou, J.; Kong, Z.; Li, N.; Yuan, Y. Germplasms, genetics and genomics for better control of disastrous wheat Fusarium head blight. Theor. Appl. Genet. 2020, 133, 1541–1568. [Google Scholar] [CrossRef] [PubMed]
- Arruda, M.P.; Lipka, A.E.; Brown, P.J.; Krill, A.M.; Thurber, C.; Brown-Guedira, G.; Dong, Y.; Foresman, B.J.; Kolb, F.L. Comparing genomic selection and marker-assisted selection for Fusarium head blight resistance in wheat (Triticum aestivum L.). Mol. Breed. 2016, 36, 84. [Google Scholar] [CrossRef]
- Lorenz, A.J.; Smith, K.P.; Jannink, J.L. Potential and optimization of genomic selection for Fusarium head blight resistance in six-row barley. Crop. Sci. 2012, 52, 1609–1621. [Google Scholar] [CrossRef]
- Galiano-Carneiro, A.L.; Boeven, P.H.G.; Maurer, H.P.; Würschum, T.; Miedaner, T. Genome-wide association study for an efficient selection of Fusarium head blight resistance in winter triticale. Euphytica 2019, 215, 4. [Google Scholar] [CrossRef]
- Rutkoski, J.; Benson, J.; Jia, Y.; Brown-Guedira, G.; Jannink, J.-L.; Sorrells, M. Evaluation of Genomic Prediction Methods for Fusarium Head Blight Resistance in Wheat. Plant. Genome 2012, 5, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Borrenpohl, D.; Huang, M.; Olson, E.; Sneller, C. The value of early-stage phenotyping for wheat breeding in the age of genomic selection. Theor. Appl. Genet. 2020, 133, 2499–2520. [Google Scholar] [CrossRef]
- Schulthess, A.W.; Zhao, Y.; Longin, C.F.H.; Reif, J.C. Advantages and limitations of multiple-trait genomic prediction for Fusarium head blight severity in hybrid wheat (Triticum aestivum L.). Theor. Appl. Genet. 2018, 131, 685–701. [Google Scholar] [CrossRef]
- Larkin, D.L.; Holder, A.L.; Mason, R.E.; Moon, D.E.; Brown-Guedira, G.; Price, P.P.; Harrison, S.A.; Dong, Y. Genome-wide analysis and prediction of Fusarium head blight resistance in soft red winter wheat. Crop. Sci. 2020, 60, 2882–2900. [Google Scholar] [CrossRef]
- Buerstmayr, M.; Buerstmayr, H. The Semidwarfing Alleles Rht-D1b and Rht-B1b Show Marked Differences in Their Associations with Anther-Retention in Wheat Heads and with Fusarium Head Blight Susceptibility. Phytopathology 2016, 106, 1544–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; He, X.; Dreisigacker, S.; He, Z.; Singh, P.K. Anther extrusion and its association with Fusarium head blight in CIMMYT wheat germplasm. Agronomy 2020, 10, 47. [Google Scholar] [CrossRef] [Green Version]
- Hamany Djande, C.Y.; Pretorius, C.; Tugizimana, F.; Piater, L.A.; Dubery, I.A. Metabolomics: A tool for cultivar phenotyping and investigation of grain crops. Agronomy 2020, 10, 831. [Google Scholar] [CrossRef]
- Kazan, K.; Gardiner, D.M. Transcriptomics of cereal–Fusarium graminearum interactions: What we have learned so far. Mol. Plant. Pathol. 2018, 19, 764–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedelsheimer, C.; Czedik-Eysenberg, A.; Grieder, C.; Lisec, J.; Technow, F.; Sulpice, R.; Altmann, T.; Stitt, M.; Willmitzer, L.; Melchinger, A.E. Genomic and metabolic prediction of complex heterotic traits in hybrid maize. Nat. Genet. 2012, 44, 217–220. [Google Scholar] [CrossRef]
- Schrag, T.A.; Westhues, M.; Schipprack, W.; Seifert, F.; Thiemann, A.; Scholten, S.; Melchinger, A.E. Beyond genomic prediction: Combining different types of omics data can improve prediction of hybrid performance in maize. Genetics 2018, 208, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Gemmer, M.R.; Richter, C.; Jiang, Y.; Schmutzer, T.; Raorane, M.L.; Junker, B.; Pillen, K.; Maurer, A. Can metabolic prediction be an alternative to genomic prediction in barley? PLoS ONE 2020, 15, e0234052. [Google Scholar] [CrossRef]
- Guo, Z.; Magwire, M.M.; Basten, C.J.; Xu, Z.; Wang, D. Evaluation of the utility of gene expression and metabolic information for genomic prediction in maize. Theor. Appl. Genet. 2016, 129, 2413–2427. [Google Scholar] [CrossRef]
- Frisch, M.; Thiemann, A.; Fu, J.; Schrag, T.A.; Scholten, S.; Melchinger, A.E. Transcriptome-based distance measures for grouping of germplasm and prediction of hybrid performance in maize. Theor. Appl. Genet. 2010, 120, 441–450. [Google Scholar] [CrossRef]
- Xu, Y.; Zhao, Y.; Wang, X.; Ma, Y.; Li, P.; Yang, Z.; Zhang, X.; Xu, C.; Xu, S. Incorporation of parental phenotypic data into multi-omic models improves prediction of yield-related traits in hybrid rice. Plant. Biotechnol. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wei, J.; Li, R.; Qu, H.; Chater, J.M.; Ma, R.; Li, Y.; Xie, W.; Jia, Z. Identification of optimal prediction models using multi-omic data for selecting hybrid rice. Heredity 2019, 123, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Westhues, M.; Schrag, T.A.; Heuer, C.; Thaller, G.; Utz, H.F.; Schipprack, W.; Thiemann, A.; Seifert, F.; Ehret, A.; Schlereth, A.; et al. Omics-based hybrid prediction in maize. Theor. Appl. Genet. 2017, 130, 1927–1939. [Google Scholar] [CrossRef] [PubMed]
- Schrauf, M.F.; Martini, J.W.R.; Simianer, H.; de los Campos, G.; Cantet, R.; Freudenthal, J.; Korte, A.; Munilla, S. Phantom Epistasis in Genomic Selection: On the Predictive Ability of Epistatic Models. G3 2020, 10, 3137–3145. [Google Scholar] [CrossRef] [PubMed]
- Weisweiler, M.; De Montaigu, A.; Ries, D.; Pfeifer, M.; Stich, B. Transcriptomic and presence/absence variation in the barley genome assessed from multi-tissue mRNA sequencing and their power to predict phenotypic traits. BMC Genom. 2019, 20, 787. [Google Scholar] [CrossRef] [PubMed]
- Westhues, M.; Heuer, C.; Thaller, G.; Fernando, R.; Melchinger, A.E. Efficient genetic value prediction using incomplete omics data. Theor. Appl. Genet. 2019, 132, 1211–1222. [Google Scholar] [CrossRef]
- Lourenco, D.; Legarra, A.; Tsuruta, S.; Masuda, Y.; Aguilar, I.; Misztal, I. Single-Step Genomic Evaluations from Theory to Practice: Using SNP Chips and Sequence Data in BLUPF90. Genes 2020, 11, 790. [Google Scholar] [CrossRef]
- Buerstmayr, H.; Steiner, B.; Lemmens, M.; Ruckenbauer, P. Resistance to Fusarium Head Blight in Winter Wheat: Heritability and Trait Associations. Crop. Sci. 2000, 40, 1012–1018. [Google Scholar] [CrossRef]
- Runcie, D.; Cheng, H. Pitfalls and remedies for cross validation with multi-trait genomic prediction methods. G3 2019, 9, 3727–3741. [Google Scholar] [CrossRef] [Green Version]
- Cullis, B.R.; Smith, A.B.; Coombes, N.E. On the design of early generation variety trials with correlated data. J. Agric. Biol. Environ. Stat. 2006, 11, 381–393. [Google Scholar] [CrossRef]
- Covarrubias-Pazaran, G. Genome-Assisted Prediction of Quantitative Traits Using the R Package sommer. PLoS ONE 2016, 11, e0156744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020.
- Rimbert, H.; Darrier, B.; Navarro, J.; Kitt, J.; Choulet, F.; Leveugle, M.; Duarte, J.; Rivière, N.; Eversole, K.; Le Gouis, J.; et al. High throughput SNP discovery and genotyping in hexaploid wheat. PLoS ONE 2018, 13, e0186329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stekhoven, D.J.; Bühlmann, P. Missforest-Non-parametric missing value imputation for mixed-type data. Bioinformatics 2012, 28, 112–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samad-Zamini, M.; Schweiger, W.; Nussbaumer, T.; Mayer, K.F.X.; Buerstmayr, H. Time-course expression QTL-atlas of the global transcriptional response of wheat to Fusarium graminearum. Plant. Biotechnol. J. 2017, 15, 1453–1464. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef]
- Appels, R.; Eversole, K.; Stein, N.; Feuillet, C.; Keller, B.; Rogers, J.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; Poland, J.; et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Endelman, J.B.; Jannink, J.-L. Shrinkage estimation of the realized relationship matrix. G3 2012, 2, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, L.; Rouseeuw, P. Finding Groups in Data: An Introduction to Cluster Analysis; Kaufman, L., Rousseeuw, P.J., Eds.; Wiley Series in Probability and Statistics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1990; ISBN 9780470316801. [Google Scholar]
- Hennig, C. Package ‘fpc’. Flexible Procedures for Clustering; University of Bologna: Bologna, Italy, 2020. [Google Scholar]
- Bustos-Korts, D.; Malosetti, M.; Chapman, S.; Biddulph, B.; van Eeuwijk, F. Improvement of Predictive Ability by Uniform Coverage of the Target Genetic Space. G3 2016, 6, 3733–3747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legarra, A.; Aguilar, I.; Misztal, I. A relationship matrix including full pedigree and genomic information. J. Dairy Sci. 2009, 92, 4656–4663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, O.; Lund, M.S. Genomic relationship matrix when some animals are not genotyped. Genet. Sel. Evol. 2010, 42, 2. [Google Scholar] [CrossRef] [Green Version]
- Amadeu, R.R.; Cellon, C.; Olmstead, J.W.; Garcia, A.A.F.; Resende, M.F.R.; Muñoz, P.R. AGHmatrix: R Package to Construct Relationship Matrices for Autotetraploid and Diploid Species: A Blueberry Example. Plant. Genome 2016, 9, 1–10. [Google Scholar] [CrossRef]
- Hänsel, H. Yield potential of barley corrected for disease infection by regression residuals. Plant. Breed. 2001, 120, 223–226. [Google Scholar] [CrossRef]
- Kugler, K.G.; Siegwart, G.; Nussbaumer, T.; Ametz, C.; Spannagl, M.; Steiner, B.; Lemmens, M.; Mayer, K.F.X.; Buerstmayr, H.; Schweiger, W. Quantitative trait loci-dependent analysis of a gene co-expression network associated with Fusarium head blight resistance in bread wheat (Triticum aestivum L.). BMC Genom. 2013, 14, 728. [Google Scholar] [CrossRef] [Green Version]
- Zenke-Philippi, C.; Thiemann, A.; Seifert, F.; Schrag, T.; Melchinger, A.E.; Scholten, S.; Frisch, M. Prediction of hybrid performance in maize with a ridge regression model employed to DNA markers and mRNA transcription profiles. BMC Genom. 2016, 17, 262. [Google Scholar] [CrossRef] [Green Version]
- Azodi, C.B.; Pardo, J.; VanBuren, R.; de Los Campos, G.; Shiu, S.H. Transcriptome-Based Prediction of Complex Traits in Maize. Plant. Cell 2020, 32, 139–151. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Li, R.; Wang, H.; Li, D.; Wang, X.; Zhang, Y.; Zhen, W.; Duan, H.; Yan, G.; Li, Y. Transcriptomics analyses reveal wheat responses to drought stress during reproductive stages under field conditions. Front. Plant. Sci. 2017, 8, 592. [Google Scholar] [CrossRef] [Green Version]
- Wagner, C.; Buerstmayr, M.; Omony, J.; Nosenko, T.; Steiner, B.; Nussbaumer, T.; Samad-Zamini, M.; Mayer, K.F.X.; Buerstmayr, H. Fusarium head blight resistance in European winter wheat: Insights from genome-wide transcriptome analysis. manuscript in preparation.
- Lamarre, S.; Frasse, P.; Zouine, M.; Labourdette, D.; Sainderichin, E.; Hu, G.; Le Berre-Anton, V.; Bouzayen, M.; Maza, E. Optimization of an RNA-seq differential gene expression analysis depending on biological replicate number and library size. Front. Plant. Sci. 2018, 9, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uzbas, F.; Opperer, F.; Sönmezer, C.; Shaposhnikov, D.; Sass, S.; Krendl, C.; Angerer, P.; Theis, F.J.; Mueller, N.S.; Drukker, M. BART-Seq: Cost-effective massively parallelized targeted sequencing for genomics, transcriptomics, and single-cell analysis. Genome Biol. 2019, 20, 155. [Google Scholar] [CrossRef] [PubMed]
- Alpern, D.; Gardeux, V.; Russeil, J.; Mangeat, B.; Meireles-Filho, A.C.A.; Breysse, R.; Hacker, D.; Deplancke, B. BRB-seq: Ultra-affordable high-throughput transcriptomics enabled by bulk RNA barcoding and sequencing. Genome Biol. 2019, 20, 1–15. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial | |||
|---|---|---|---|
| Year | FS | FC | FG |
| 2011 | 26 (2) | 26 (4) | |
| 2012 | 58 (2) | 58 (2) | |
| 2013 | 96 (2) | 96 (2) | |
| 2014 | 96 (2) | ||
| 2015 | 96 (2) | 96 (2) | |
| Set | Trait | Min | Mean | Max | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| All trials | FHB | 51.89 | 114.62 | 282.47 | 2338.49 | 0.00 | 629.45 | 10226.20 | 0.18 | 0.94 |
| AR | 0.00 | 52.96 | 93.13 | 734.03 | 68.81 | 22.42 | 156.29 | 0.75 | 0.97 | |
| PH | 65.71 | 78.23 | 104.68 | 72.36 | 3.74 | 0.00 | 10.84 | 0.83 | 0.98 | |
| AD | 20.20 | 24.67 | 30.00 | 5.62 | 0.85 | 0.00 | 1.36 | 0.72 | 0.97 | |
| Isolates † | FHBFS | 0.00 | 39.40 | 177.92 | 1308.28 | 335.18 | 831.78 | 0.53 | 0.91 | |
| FHBFC | 17.04 | 227.36 | 846.55 | 35605.89 | 3776.73 | 6610.13 | 0.77 | 0.95 | ||
| FHBFG | 69.34 | 201.10 | 452.40 | 7572.85 | 1877.78 | 5436.90 | 0.51 | 0.87 | ||
| Ind. Sel. ‡ | ARwoFS | 0.00 | 50.07 | 91.55 | 744.94 | 47.46 | 47.46 | 160.38 | 0.74 | 0.97 |
| ARwoFC | 0.00 | 50.55 | 84.34 | 568.15 | 68.44 | 24.75 | 174.89 | 0.68 | 0.93 | |
| ARwoFG | 0.00 | 54.45 | 93.73 | 782.06 | 42.69 | 42.69 | 129.45 | 0.78 | 0.97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michel, S.; Wagner, C.; Nosenko, T.; Steiner, B.; Samad-Zamini, M.; Buerstmayr, M.; Mayer, K.; Buerstmayr, H. Merging Genomics and Transcriptomics for Predicting Fusarium Head Blight Resistance in Wheat. Genes 2021, 12, 114. https://doi.org/10.3390/genes12010114
Michel S, Wagner C, Nosenko T, Steiner B, Samad-Zamini M, Buerstmayr M, Mayer K, Buerstmayr H. Merging Genomics and Transcriptomics for Predicting Fusarium Head Blight Resistance in Wheat. Genes. 2021; 12(1):114. https://doi.org/10.3390/genes12010114
Chicago/Turabian StyleMichel, Sebastian, Christian Wagner, Tetyana Nosenko, Barbara Steiner, Mina Samad-Zamini, Maria Buerstmayr, Klaus Mayer, and Hermann Buerstmayr. 2021. "Merging Genomics and Transcriptomics for Predicting Fusarium Head Blight Resistance in Wheat" Genes 12, no. 1: 114. https://doi.org/10.3390/genes12010114
APA StyleMichel, S., Wagner, C., Nosenko, T., Steiner, B., Samad-Zamini, M., Buerstmayr, M., Mayer, K., & Buerstmayr, H. (2021). Merging Genomics and Transcriptomics for Predicting Fusarium Head Blight Resistance in Wheat. Genes, 12(1), 114. https://doi.org/10.3390/genes12010114