The Yin and Yang-Like Clinical Implications of the CDKN2A/ARF/CDKN2B Gene Cluster in Acute Lymphoblastic Leukemia


Abstract
:1. Introduction
2. Genetic and Epigenetic View of the CDKN2A/B Gene Cluster in ALL
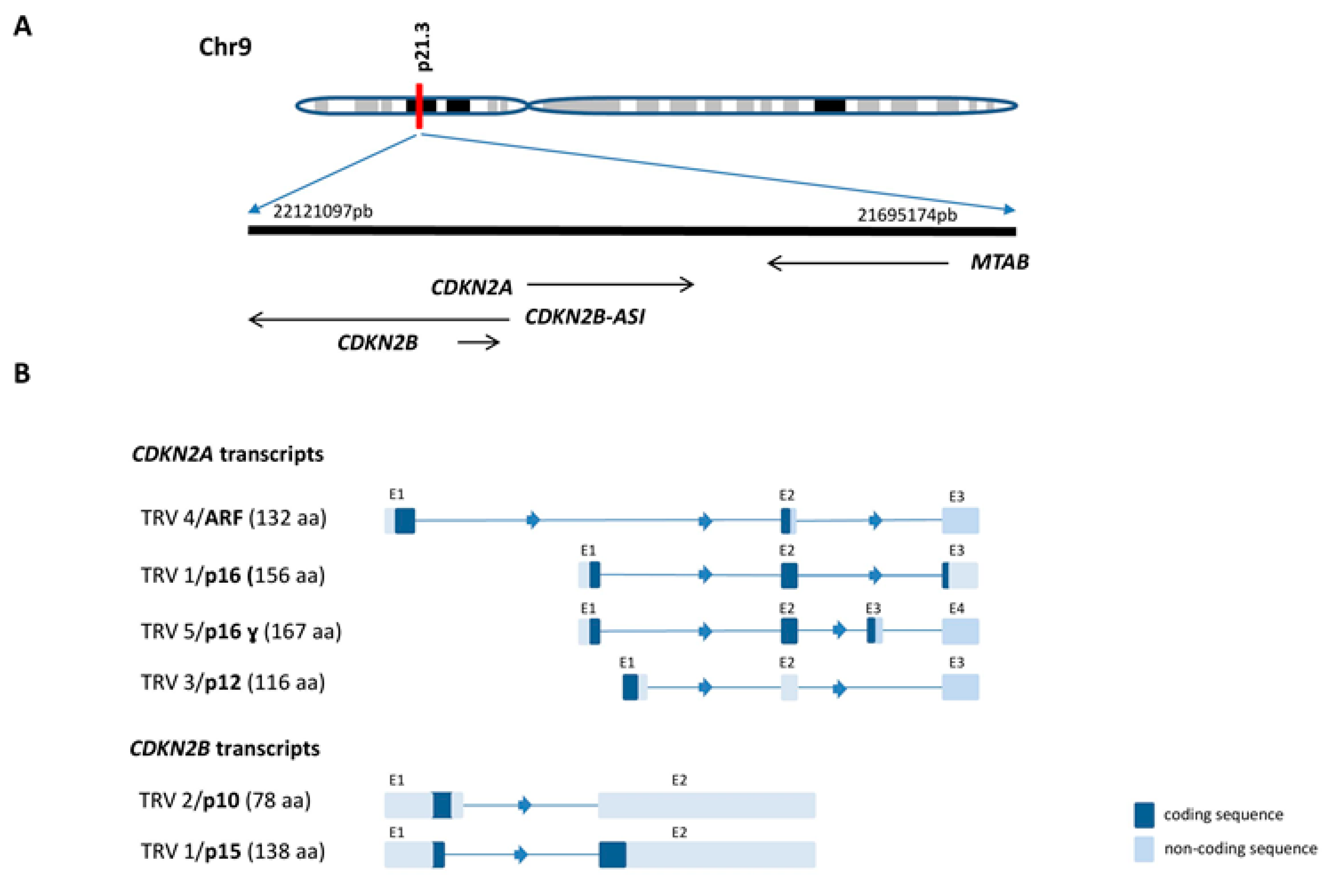
2.1. CDKN2A/B Gene Cluster Organization and Transcripts
2.2. 9p21.3 Deletion in ALL
2.2.1. Methods to Detect the Alteration and Possible Origin
2.2.2. del(9p21.3) in BCP-ALL
2.2.3. del(9p21.3) in T-ALL
2.3. Epigenetic Modifications at the CDKN2A/B Gene Promoter (T-ALL and BCP-ALL)
2.4. Germline Predisposition Variants in the CDKN2A/B Gene Cluster (T-ALL and BCP-ALL)
3. Clinical Impact of CDKN2A/B Alterations in ALL
3.1. Clinical Implications of Deletions in BCP-ALL
3.2. Clinical Implications of Deletions in T-ALL
3.3. Clinical Impact of Epigenetic Modifications (BCP-ALL and T-ALL)
4. Functional Implications of the CDKN2A/B Locus in ALL
4.1. Role of INK4a/ARF Proteins in Leukemogenesis
4.2. Role of the INK4a/ARF Proteins in Genomic Instability
4.3. Consequences of Germline Mutations
5. Implications of the CDKN2A/B Gene Cluster for Treatment Resistance/Relapse
6. Therapeutic Approaches to Targeting the INK4 Tumor-Suppressor Protein Family
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pui, C.H.; Campana, D.; Pei, D.; Bowman, W.P.; Sandlund, J.T.; Kaste, S.C.; Ribeiro, R.C.; Rubnitz, J.E.; Raimondi, S.C.; Onciu, M.; et al. Treating Childhood Acute Lymphoblastic Leukemia without Cranial Irradiation. N. Engl. J. Med. 2009, 360, 2730–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, J.M.; Buck, G.; Burnett, A.K.; Chopra, R.; Wiernik, P.H.; Richards, S.M.; Lazarus, H.M.; Franklin, I.M.; Litzow, M.R.; Ciobanu, N.; et al. Induction therapy for adults with acute lymphoblastic leukemia: Results of more than 1500 patients from the international ALL trial: MRC UKALL XII/ECOG E2993. Blood 2005, 106, 3760–3767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Ouelle, D.E.; Zindy, F.; Ashmun, R.A.; Sherr, C.J. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 1995, 83, 993–1000. [Google Scholar] [CrossRef] [Green Version]
- Sharpless, N.E.; Depinho, R.A. The INK4A/ARF locus and its two gene products. Curr. Opin. Genet. Dev. 1999, 9, 22–30. [Google Scholar] [CrossRef]
- Sherr, C.J. The INK4a/ARF network in tumour suppression. Nat. Rev. Mol. Cell Biol. 2001, 2, 731–737. [Google Scholar] [CrossRef]
- Hannon, G.J.; Beach, D. pl5INK4B is a potentia| effector of TGF-β-induced cell cycle arrest. Nature 1994, 371, 257–261. [Google Scholar] [CrossRef]
- Lin, Y.C.; Diccianni, M.B.; Kim, Y.; Lin, H.H.; Lee, C.H.; Lin, R.J.; Joo, S.H.; Li, J.; Chuang, T.J.; Yang, A.S.; et al. Human p16γ, a novel transcriptional variant of p16INK4A, coexpresses with p16INK4A in cancer cells and inhibits cell-cycle progression. Oncogene 2007, 26, 7017–7027. [Google Scholar] [CrossRef] [Green Version]
- Robertson, K.D.; Jones, P.A. Tissue-specific alternative splicing in the human INK4a/ARF cell cycle regulatory locus. Oncogene 1999, 18, 3810–3820. [Google Scholar] [CrossRef] [Green Version]
- Tsubari, M.; Tiihonen, E.; Laiho, M. Cloning and characterization of p10, an alternatively spliced form of p15 cyclin-dependent kinase inhibitor. Cancer Res. 1997, 57, 2966–2973. [Google Scholar]
- Georgopoulos, K. The making of a lymphocyte: The choice among disparate cell fates and the IKAROS enigma. Genes Dev. 2017, 31, 439–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raschke, S.; Balz, V.; Efferth, T.; Schulz, W.A.; Florl, A.R. Homozygous deletions of CDKN2A caused by alternative mechanisms in various human cancer cell lines. Genes Chromosom. Cancer 2005, 42, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, Y.; Inoue, K.; Sasaki, S.; Hayashi, Y.; Matsuo, Y.; Lieber, M.R.; Mizoguchi, H.; Yokota, J.; Kohno, T. Prevalent involvement of illegitimate V(D)J recombination in chromosome 9p21 deletions in lymphoid leukemia. J. Biol. Chem. 2002, 277, 46289–46297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novara, F.; Beri, S.; Bernardo, M.E.; Bellazzi, R.; Malovini, A.; Ciccone, R.; Cometa, A.M.; Locatelli, F.; Giorda, R.; Zuffardi, O. Different molecular mechanisms causing 9p21 deletions in acute lymphoblastic leukemia of childhood. Hum. Genet. 2009, 126, 511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaemmanuil, E.; Rapado, I.; Li, Y.; Potter, N.E.; Wedge, D.C.; Tubio, J.; Alexandrov, L.B.; Van Loo, P.; Cooke, S.L.; Marshall, J.; et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat. Genet. 2014, 46, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Cayuela, J.M.; Gardie, B.; Sigaux, F. Disruption of the multiple tumor suppressor gene MTS1/p16(INK4a)/CDKN2 by illegitimate V(D)J recombinase activity in T-cell acute lymphoblastic leukemias. Blood 1997, 90, 3720–3726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morison, I.M.; Ellis, L.M.; Teague, L.R.; Reeve, A.E. Preferential loss of maternal 9p alleles in childhood acute lymphoblastic leukemia. Blood 2002, 99, 375–377. [Google Scholar] [CrossRef] [Green Version]
- Iacobucci, I.; Ferrari, A.; Lonetti, A.; Papayannidis, C.; Paoloni, F.; Trino, S.; Storlazzi, C.T.; Ottaviani, E.; Cattina, F.; Impera, L.; et al. CDKN2A/B alterations impair prognosis in adult BCR-ABL1-positive acute lymphoblastic leukemia patients. Clin. Cancer Res. 2011, 17, 7413–7423. [Google Scholar] [CrossRef] [Green Version]
- Braun, M.; Pastorczak, A.; Fendler, W.; Madzio, J.; Tomasik, B.; Taha, J.; Bielska, M.; Sedek, L.; Szczepanski, T.; Matysiak, M.; et al. Biallelic loss of CDKN2A is associated with poor response to treatment in pediatric acute lymphoblastic leukemia. Leuk. Lymphoma 2017, 58, 1162–1171. [Google Scholar] [CrossRef]
- Schwab, C.J.; Chilton, L.; Morrison, H.; Jones, L.; Al-Shehhi, H.; Erhorn, A.; Russell, L.J.; Moorman, A.V.; Harrison, C.J. Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: Association with cytogenetics and clinical features. Haematologica 2013, 98, 1081–1088. [Google Scholar] [CrossRef]
- Paulsson, K.; Cazier, J.B.; MacDougall, F.; Stevens, J.; Stasevich, I.; Vrcelj, N.; Chaplin, T.; Lillington, D.M.; Lister, T.A.; Young, B.D. Microdeletions are a general feature of adult and adolescent acute lymphoblastic leukemia: Unexpected similarities with pediatric disease. Proc. Natl. Acad. Sci. USA 2008, 105, 6708–6713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, R.; Ogawa, S.; Nowak, D.; Kawamata, N.; Akagi, T.; Kato, M.; Sanada, M.; Weiss, T.; Haferlach, C.; Dugas, M.; et al. Genomic profiling of adult acute lymphoblastic leukemia by single nucleotide polymorphism oligonucleotide microarray and comparison to pediatric acute lymphoblastic leukemia. Haematologica 2010, 95, 1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullighan, C.G.; Miller, C.B.; Radtke, I.; Phillips, L.A.; Dalton, J.; Ma, J.; White, D.; Hughes, T.P.; Le Beau, M.M.; Pui, C.H.; et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 2008, 453, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Sulong, S.; Moorman, A.V.; Irving, J.A.; Strefford, J.C.; Konn, Z.J.; Case, M.C.; Minto, L.; Barber, K.E.; Parker, H.; Wright, S.L.; et al. A comprehensive analysis of the CDKN2A gene in childhood acute lymphoblastic leukemia reveals genomic deletion, copy number neutral loss of heterozygosity, and association with specific cytogenetic subgroups. Blood 2009, 113, 100–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, H.; Raum, K.; Markovic, S.; Nowak, V.; Fey, S.; Obländer, J.; Pressler, J.; Böhm, V.; Brüggemann, M.; Wunderle, L.; et al. Genomic CDKN2A/2B deletions in adult Ph+ ALL are adverse despite allogeneic stem cell transplantation. Blood 2018, 131, 1464–1475. [Google Scholar] [CrossRef]
- Steeghs, E.M.; Boer, J.M.; Hoogkamer, A.Q.; Boeree, A.; De Haas, V.; De Groot-Kruseman, H.A.; Horstmann, M.A.; Escherich, G.; Pieters, R.; Den Boer, M.L. Copy number alterations in B-cell development genes, drug resistance, and clinical outcome in pediatric B-cell precursor acute lymphoblastic leukemia. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Ensor, H.M.; Schwab, C.; Russell, L.J.; Richards, S.M.; Morrison, H.; Masic, D.; Jones, L.; Kinsey, S.E.; Vora, A.J.; Mitchell, C.D.; et al. Demographic, clinical, and outcome features of children with acute lymphoblastic leukemia and CRLF2 deregulation: Results from the MRC ALL97 clinical trial. Blood 2011, 117, 2129–2136. [Google Scholar] [CrossRef]
- Zaliova, M.; Potuckova, E.; Hovorkova, L.; Musilova, A.; Winkowska, L.; Fiser, K.; Stuchly, J.; Mejstrikova, E.; Starkova, J.; Zuna, J.; et al. ERG deletions in childhood acute lymphoblastic leukemia with DUX4 rearrangements are mostly polyclonal, prognostically relevant and their detection rate strongly depends on screening method sensitivity. Haematologica 2019, 104, 1407–1416. [Google Scholar] [CrossRef] [Green Version]
- Russell, L.J.; Akasaka, T.; Majid, A.; Sugimoto, K.J.; Loraine Karran, E.; Nagel, I.; Harder, L.; Claviez, A.; Gesk, S.; Moorman, A.V.; et al. t(6;14)(p22;q32): A new recurrent IGH@ translocation involving ID4 in B-cell precursor acute lymphoblastic leukemia (BCP-ALL). Blood 2008, 111, 387–391. [Google Scholar] [CrossRef] [Green Version]
- Passet, M.; Boissel, N.; Sigaux, F.; Saillard, C.; Bargetzi, M.; Ba, I.; Thomas, X.; Graux, C.; Chalandon, Y.; Leguay, T.; et al. PAX5 P80R mutation identifies a novel subtype of B-cell precursor acute lymphoblastic leukemia with favorable outcome. Blood 2019, 133, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019, 51, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Zhang, J.; Harvey, R.C.; Collins-Underwood, J.R.; Schulman, B.A.; Phillips, L.A.; Tasian, S.K.; Loh, M.L.; Su, X.; Liu, W.; et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2009, 106, 9414–9418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheijen, B.; Boer, J.M.; Marke, R.; Tijchon, E.; Van Ingen Schenau, D.; Waanders, E.; Van Emst, L.; Van Der Meer, L.T.; Pieters, R.; Escherich, G.; et al. Tumor suppressors BTG1 and IKZF1 cooperate during mouse leukemia development and increase relapse risk in B-cell precursor acute lymphoblastic leukemia patients. Haematologica 2017, 102, 541–551. [Google Scholar] [CrossRef] [Green Version]
- Kathiravan, M.; Singh, M.; Bhatia, P.; Trehan, A.; Varma, N.; Sachdeva, M.S.; Bansal, D.; Jain, R.; Naseem, S. Deletion of CDKN2A/B is associated with inferior relapse free survival in pediatric B cell acute lymphoblastic leukemia. Leuk. Lymphoma 2019, 60, 433–441. [Google Scholar] [CrossRef]
- Heerema, N.A.; Nachman, J.B.; Sather, H.N.; Sensel, M.G.; Lee, M.K.; Hutchinson, R.; Lange, B.J.; Steinherz, P.G.; Bostrom, B.; Gaynon, P.S.; et al. Hypodiploidy with less than 45 chromosomes confers adverse risk in childhood acute lymphoblastic leukemia: A report from the children’s cancer group. Blood 1999, 12, 4036–4045. [Google Scholar]
- Moreno, T.C.; Gustafsson, G.; Garwicz, S.; Grander, D.; Jonmundsson, G.K.; Frost, B.M.; Mäkipernaa, A.; Rasool, O.; Savolainen, E.R.; Schmiegelow, K.; et al. Deletion of the Ink4-locus (the p16ink4a, p14ARF and p15ink4b genes) predicts relapse in children with ALL treated according to the Nordic protocols NOPHO-86 and NOPHO-92. Leukemia 2002, 16, 2037–2045. [Google Scholar] [CrossRef] [Green Version]
- van Zutven, L.J.; van Drunen, E.; de Bont, J.M.; Wattel, M.M.; Den Boer, M.L.; Pieters, R.; Hagemeijer, A.; Slater, R.M.; Beverloo, H.B. CDKN2 deletions have no prognostic value in childhood precursor-B acute lymphoblastic leukaemia. Leukemia 2005, 19, 1281–1284. [Google Scholar] [CrossRef] [Green Version]
- Mirebeau, D.; Acquaviva, C.; Suciu, S.; Bertin, R.; Dastugue, N.; Robert, A.; Boutard, P.; Méchinaud, F.; Plouvier, E.; Otten, J.; et al. The prognostic significance of CDKN2A, CDKN2B and MTAP inactivation in B-lineage acute lymphoblastic leukemia of childhood. Results of the EORTC studies 58881 and 58951. Haematologica 2006, 7, 881–885. [Google Scholar]
- Mullighan, C.G.; Su, X.; Zhang, J.; Radtke, I.; Phillips, L.A.; Miller, C.B.; Ma, J.; Liu, W.; Cheng, C.; Schulman, B.A.; et al. Deletion of IKZF1 and Prognosis in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2009, 360, 470–480. [Google Scholar] [CrossRef]
- Kuchinskaya, E.; Heyman, M.; Nordgren, A.; Söderhäll, S.; Forestier, E.; Wehner, P.; Vettenranta, K.; Jonsson, O.; Wesenberg, F.; Sahlén, S.; et al. Interphase fluorescent in situ hybridization deletion analysis of the 9p21 region and prognosis in childhood acute lymphoblastic leukaemia (ALL): Results from a prospective analysis of 519 Nordic patients treated according to the NOPHO-ALL 2000 protocol. Br. J. Haematol. 2011, 152, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Krentz, S.; Hof, J.; Mendioroz, A.; Vaggopoulou, R.; Dörge, P.; Lottaz, C.; Engelmann, J.C.; Groeneveld, T.W.L.; Körner, G.; Seeger, K.; et al. Prognostic value of genetic alterations in children with first bone marrow relapse of childhood B-cell precursor acute lymphoblastic leukemia. Leukemia 2013, 27, 295–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forero-Castro, M.; Robledo, C.; Benito, R.; Abáigar, M.; África Martín, A.; Arefi, M.; Fuster, J.L.; de Las Heras, N.; Rodríguez, J.N.; Quintero, J.; et al. Genome-Wide DNA Copy Number Analysis of Acute Lymphoblastic Leukemia Identifies New Genetic Markers Associated with Clinical Outcome. PLoS ONE 2016, 11, e0148972. [Google Scholar] [CrossRef] [PubMed]
- Messina, M.; Chiaretti, S.; Fedullo, A.L.; Piciocchi, A.; Puzzolo, M.C.; Lauretti, A.; Gianfelici, V.; Apicella, V.; Fazi, P.; Te Kronnie, G.; et al. Clinical significance of recurrent copy number aberrations in B-lineage acute lymphoblastic leukaemia without recurrent fusion genes across age cohorts. Br. J. Haematol. 2017, 178, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Sutton, R.; Venn, N.C.; Law, T.; Boer, J.M.; Trahair, T.N.; Ng, A.; Den Boer, M.L.; Dissanayake, A.; Giles, J.E.; Dalzell, P.; et al. A risk score including microdeletions improves relapse prediction for standard and medium risk precursor B-cell acute lymphoblastic leukaemia in children. Br. J. Haematol. 2018, 180, 550–562. [Google Scholar] [CrossRef]
- Parker, C.; Krishnan, S.; Hamadeh, L.; Irving, J.A.; Kuiper, R.P.; Révész, T.; Hoogerbrugge, P.; Hancock, J.; Sutton, R.; Moorman, A.V.; et al. Outcomes of patients with childhood B-cell precursor acute lymphoblastic leukaemia with late bone marrow relapses: Long-term follow-up of the ALLR3 open-label randomised trial. Lancet Haematol. 2019, 6, e204–e216. [Google Scholar] [CrossRef] [Green Version]
- Mullighan, C.G.; Goorha, S.; Radtke, I.; Miller, C.B.; Coustan-Smith, E.; Dalton, J.D.; Girtman, K.; Mathew, S.; Ma, J.; Pounds, S.B.; et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007, 446, 758–764. [Google Scholar] [CrossRef]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Karrman, K.; Castor, A.; Behrendtz, M.; Forestier, E.; Olsson, L.; Ehinger, M.; Biloglav, A.; Fioretos, T.; Paulsson, K.; Johansson, B. Deep sequencing and SNP array analyses of pediatric T-cell acute lymphoblastic leukemia reveal NOTCH1 mutations in minor subclones and a high incidence of uniparental isodisomies affecting CDKN2A. J. Hematol. Oncol. 2015, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Vicente, C.; Schwab, C.; Broux, M.; Geerdens, E.; Degryse, S.; Demeyer, S.; Lahortiga, I.; Elliott, A.; Chilton, L.; La Starza, R.; et al. Targeted sequencing identifies associations between IL7R-JAK mutations and epigenetic modulators in T-cell acute lymphoblastic leukemia. Haematologica 2015, 100, 1301–1310. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, T.C.; Liang, D.C.; Liu, H.C.; Jaing, T.H.; Chen, S.H.; Hou, J.Y.; Yang, C.P.; Huang, Y.J.; Yao, H.W.; Huang, T.Y.; et al. Clinical and biological relevance of genetic alterations in pediatric T-cell acute lymphoblastic leukemia in Taiwan. Pediatr. Blood Cancer 2019, 66, e27496. [Google Scholar] [CrossRef] [PubMed]
- Noronha, E.P.; Codeço Marques, L.V.; Andrade, F.G.; Santos Thuler, L.C.; Terra-Granado, E.; Pombo-De-Oliveira, M.S. The profile of immunophenotype and genotype aberrations in subsets of pediatric T-cell acute lymphoblastic leukemia. Front. Oncol. 2019, 9, 316. [Google Scholar] [CrossRef] [PubMed]
- Thakral, D.; Kaur, G.; Gupta, R.; Benard, A.; Savola, S.; Singh, I.K.; Anand, R.; Rani, L.; Verma, P.; Vashishtha, S.; et al. Rapid Identification of Key Copy Number Alterations in B- and T-Cell Acute Lymphoblastic Leukemia by Digital Multiplex Ligation-Dependent Probe Amplification. Front. Oncol. 2019, 9, 871. [Google Scholar] [CrossRef] [Green Version]
- Moorman, A.V.; Harrison, C.J.; Buck, G.A.; Richards, S.M.; Secker-Walker, L.M.; Martineau, M.; Vance, G.H.; Cherry, A.M.; Higgins, R.R.; Fielding, A.K.; et al. Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): Analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood 2007, 109, 3189–3197. [Google Scholar]
- Nahi, H.; Hägglund, H.; Ahlgren, T.; Bernell, P.; Hardling, M.; Karlsson, K.; Lazarevic, V.L.; Linderholm, M.; Smedmyr, B.; Åström, M.; et al. An investigation into whether deletions in 9p reflect prognosis in adult precursor B-cell acute lymphoblastic leukemia: a multi-center study of 381 patients. Haematologica 2008, 93, 1734–1738. [Google Scholar] [CrossRef] [Green Version]
- Yanada, M.; Takeuchi, J.; Sugiura, I.; Akiyama, H.; Usui, N.; Yagasaki, F.; Nishii, K.; Ueda, Y.; Takeuchi, M.; Miyawaki, S.; et al. Karyotype at diagnosis is the major prognostic factor predicting relapse-free survival for patients with Philadelphia chromosome-positive acute lymphoblastic leukemia treated with imatinib-combined chemotherapy. Haematologica 2008, 93, 287–290. [Google Scholar] [CrossRef]
- Moorman, A.V.; Schwab, C.; Ensor, H.M.; Russell, L.J.; Morrison, H.; Jones, L.; Masic, D.; Patel, B.; Rowe, J.M.; Tallman, M.; et al. IGH@ translocations, CRLF2 deregulation, and microdeletions in adolescents and adults with acute lymphoblastic leukemia. J. Clin. Oncol. 2012, 30, 3100–3108. [Google Scholar] [CrossRef]
- Ribera, J.; Morgades, M.; Zamora, L.; Montesinos, P.; Gómez-Seguí, I.; Pratcorona, M.; Sarrà, J.; Guàrdia, R.; Nomdedeu, J.; Tormo, M.; et al. Prognostic significance of copy number alterations in adolescent and adult patients with precursor B acute lymphoblastic leukemia enrolled in PETHEMA protocols. Cancer 2015, 121, 3809–3817. [Google Scholar] [CrossRef] [Green Version]
- Xu, N.; Li, Y.L.; Zhou, X.; Cao, R.; Li, H.; Lu, Q.S.; Li, L.; Lu, Z.Y.; Huang, J.X.; Sun, J.; et al. CDKN2 Gene Deletion as Poor Prognosis Predictor Involved in the Progression of Adult B-Lineage Acute Lymphoblastic Leukemia Patients. J. Cancer 2015, 6, 1114. [Google Scholar] [CrossRef] [Green Version]
- Xu, N.; Li, Y.L.; Li, X.; Zhou, X.; Cao, R.; Li, H.; Li, L.; Lu, Z.Y.; Huang, J.X.; Fan, Z.P.; et al. Correlation between deletion of the CDKN2 gene and tyrosine kinase inhibitor resistance in adult Philadelphia chromosome-positive acute lymphoblastic leukemia. J. Hematol. Oncol. 2016, 9, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seol, C.A.; Cho, Y.U.; Jang, S.; Park, C.J.; Lee, J.H.; Lee, J.H.; Lee, K.H.; Seo, E.J. Prognostic significance of recurrent additional chromosomal abnormalities in adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Cancer Genet. 2017, 216, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Mason, C.C.; Glenn, M.J.; Paxton, C.N.; South, S.T.; Cessna, M.H.; Asch, J.; Cobain, E.F.; Bixby, D.L.; Smith, L.B.; et al. Genomic analysis of adult B-ALL identifies potential markers of shorter survival. Leuk. Res. 2017, 56, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Kantarjian, H.M.; Sasaki, K.; Ravandi, F.; Ko, H.; Cameron Yin, C.; Garcia-Manero, G.; Cortes, J.E.; Garris, R.; O’Brien, S.M.; et al. Outcomes associated with +der(22)t(9;22) and −9/9p in patients with Philadelphia chromosome-positive acute lymphoblastic leukemia receiving chemotherapy plus a tyrosine kinase inhibitor. Am. J. Hematol. 2017, 92, 238–243. [Google Scholar] [CrossRef]
- Lafage-Pochitaloff, M.; Baranger, L.; Hunault, M.; Cuccuini, W.; Lefebvre, C.; Bidet, A.; Tigaud, I.; Eclache, V.; Delabesse, E.; Bilhou-Nabéra, C.; et al. Impact of cytogenetic abnormalities in adults with Ph-negative B-cell precursor acute lymphoblastic leukemia. Blood 2017, 130, 1832–1844. [Google Scholar] [CrossRef] [Green Version]
- Fedullo, A.L.; Messina, M.; Elia, L.; Piciocchi, A.; Gianfelici, V.; Lauretti, A.; Soddu, S.; Puzzolo, M.C.; Minotti, C.; Ferrara, F.; et al. Prognostic implications of additional genomic lesions in adult Philadelphia chromosome-positive acute lymphoblastic leukemia. Haematologica 2019, 104, 312–318. [Google Scholar] [CrossRef]
- Ribera, J.; Zamora, L.; Morgades, M.; Vives, S.; Granada, I.; Montesinos, P.; Gómez-Seguí, I.; Mercadal, S.; Guàrdia, R.; Nomdedeu, J.; et al. Molecular profiling refines minimal residual disease-based prognostic assessment in adults with Philadelphia chromosome-negative B-cell precursor acute lymphoblastic leukemia. Genes Chromosom. Cancer 2019, 58, 815–819. [Google Scholar] [CrossRef]
- Sánchez, R.; Ribera, J.; Morgades, M.; Ayala, R.; Onecha, E.; Ruiz-Heredia, Y.; Juárez-Rufián, A.; de Nicolás, R.; Sánchez-Pina, J.; Vives, S.; et al. A novel targeted RNA-Seq panel identifies a subset of adult patients with acute lymphoblastic leukemia with BCR-ABL1-like characteristics. Blood Cancer J. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Marks, D.I.; Paietta, E.M.; Moorman, A.V.; Richards, S.M.; Buck, G.; DeWald, G.; Ferrando, A.; Fielding, A.K.; Goldstone, A.H.; Ketterling, R.P.; et al. T-cell acute lymphoblastic leukemia in adults: Clinical features, immunophenotype, cytogenetics, and outcome from the large randomized prospective trial (UKALL XII/ECOG 2993). Blood 2009, 114, 5136–5145. [Google Scholar] [CrossRef] [Green Version]
- Grossmann, V.; Haferlach, C.; Weissmann, S.; Roller, A.; Schindela, S.; Poetzinger, F.; Stadler, K.; Bellos, F.; Kern, W.; Haferlach, T.; et al. The molecular profile of adult T-cell acute lymphoblastic leukemia: Mutations in RUNX1 and DNMT3A are associated with poor prognosis in T-ALL. Genes Chromosom. Cancer 2013, 52, 410–422. [Google Scholar] [CrossRef]
- Van Vlierberghe, P.; Ambesi-Impiombato, A.; De Keersmaecker, K.; Hadler, M.; Paietta, E.; Tallman, M.S.; Rowe, J.M.; Forne, C.; Rue, M.; Ferrando, A.A. Prognostic relevance of integrated genetic profiling in adult T-cell acute lymphoblastic leukemia. Blood 2013, 122, 74–82. [Google Scholar] [PubMed]
- Dirse, V.; Bertasiute, A.; Gineikiene, E.; Zvirblis, T.; Dambrauskiene, R.; Gerbutavicius, R.; Juozaityte, E.; Malciute, L.; Paulsson, K.; Griskevicius, L. A population-based single nucleotide polymorphism array analysis of genomic aberrations in younger adult acute lymphoblastic leukemia patients. Genes Chromosom. Cancer 2015, 54, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Yuan, T.; Li, Y.; Feng, J.; Gong, X.; Li, Q.; Zhao, X.; Liu, K.; Tang, K.; Tian, Z.; et al. Prognostic significance of copy number alterations detected by multi‑link probe amplification of multiple genes in adult acute lymphoblastic leukemia. Oncol. Lett. 2018, 15, 5359–5367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genescà, E.; Lazarenkov, A.; Morgades, M.; Berbis, G.; Ruíz-Xivillé, N.; Gómez-Marzo, P.; Ribera, J.; Juncà, J.; González-Pérez, A.; Mercadal, S.; et al. Frequency and clinical impact of CDKN2A/ARF/CDKN2B gene deletions as assessed by in-depth genetic analyses in adult T cell acute lymphoblastic leukemia. J. Hematol. Oncol. 2018, 11, 1–4. [Google Scholar]
- Jang, W.; Park, J.; Kwon, A.; Choi, H.; Kim, J.; Lee, G.D.; Han, E.; Jekarl, D.W.; Chae, H.; Han, K.; et al. CDKN2B downregulation and other genetic characteristics in T-acute lymphoblastic leukemia. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Notta, F.; Mullighan, C.G.; Wang, J.C.; Poeppl, A.; Doulatov, S.; Phillips, L.A.; Ma, J.; Minden, M.D.; Downing, J.R.; Dick, J.E. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature 2011, 469, 362. [Google Scholar] [CrossRef]
- Schmitz, M.; Breithaupt, P.; Scheidegger, N.; Cario, G.; Bonapace, L.; Meissner, B.; Mirkowska, P.; Tchinda, J.; Niggli, F.K.; Stanulla, M.; et al. Xenografts of highly resistant leukemia recapitulate the clonal composition of the leukemogenic compartment. Blood 2011, 118, 1854–1864. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Phillips, L.A.; Su, X.; Ma, J.; Miller, C.B.; Shurtleff, S.A.; Downing, J.R. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 2008, 322, 1377–1380. [Google Scholar]
- van Delft, F.W.; Horsley, S.; Colman, S.; Anderson, K.; Bateman, C.; Kempski, H.; Zuna, J.; Eckert, C.; Saha, V.; Kearney, L.; et al. Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood 2011, 117, 6247–6254. [Google Scholar]
- Ribera, J.; Zamora, L.; Morgades, M.; Mallo, M.; Solanes, N.; Batlle, M.; Vives, S.; Granada, I.; Juncà, J.; Malinverni, R.; et al. Copy number profiling of adult relapsed B-cell precursor acute lymphoblastic leukemia reveals potential leukemia progression mechanisms. Genes Chromosom. Cancer 2017, 56, 810–820. [Google Scholar]
- Hebert, J.; Cayuela, J.M.; Berkeley, J.; Sigaux, F. Candidate tumor-suppressor genes MTS1 (p16INK4A) and MTS2 (p15INK4B) display frequent homozygous deletions in primary cells from T- but not from B-cell lineage acute lymphoblastic leukemias. Blood 1994, 84, 4038–4044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitale, A. Adult T-cell acute lymphoblastic leukemia: Biologic profile at presentation and correlation with response to induction treatment in patients enrolled in the GIMEMA LAL 0496 protocol. Blood 2006, 107, 473–479. [Google Scholar] [PubMed] [Green Version]
- Studniak, E.; Maloney, E.; Ociepa, T.; Urasiński, T.; Skonieczka, K.; Haus, O.; Poluha, A.; Kowalczyk, J.; Zajączek, S. Allelic loss of selected tumor suppressor genes in acute lymphoblastic leukemia in children. Polish J. Pathol. 2013, 64, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Coustan-Smith, E.; Mullighan, C.G.; Onciu, M.; Behm, F.G.; Raimondi, S.C.; Pei, D.; Cheng, C.; Su, X.; Rubnitz, J.E.; Basso, G.; et al. Early T-cell precursor leukaemia: A subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009, 10, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Ferrando, A.A.; Neuberg, D.S.; Staunton, J.; Loh, M.L.; Huard, C.; Raimondi, S.C.; Behm, F.G.; Pui, C.H.; Downing, J.R.; Gilliland, D.G.; et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002, 1, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Homminga, I.; Pieters, R.; Langerak, A.W.; de Rooi, J.J.; Stubbs, A.; Verstegen, M.; Vuerhard, M.; Buijs-Gladdines, J.; Kooi, C.; Klous, P.; et al. Integrated Transcript and Genome Analyses Reveal NKX2-1 and MEF2C as Potential Oncogenes in T Cell Acute Lymphoblastic Leukemia. Cancer Cell 2011, 19, 484–497. [Google Scholar] [CrossRef]
- La Starza, R.; Borga, C.; Barba, G.; Pierini, V.; Schwab, C.; Matteucci, C.; Lema Fernandez, A.G.; Leszl, A.; Cazzaniga, G.; Chiaretti, S.; et al. Genetic profile of T-cell acute lymphoblastic leukemias with MYC translocations. Blood 2014, 124, 3577–3582. [Google Scholar] [CrossRef]
- Milani, G.; Matthijssens, F.; Van Loocke, W.; Durinck, K.; Roels, J.; Peirs, S.; Thénoz, M.; Pieters, T.; Reunes, L.; Lintermans, B.; et al. Genetic characterization and therapeutic targeting of MYC -rearranged T cell acute lymphoblastic leukaemia. Br. J. Haematol. 2019, 185, 169–174. [Google Scholar]
- Herman, J.G.; Civin, C.I.; Issa, J.P.J.; Collector, M.I.; Sharkis, S.J.; Baylin, S.B. Distinct patterns of inactivation of p15INK4B and p16INK4A characterize the major types of hematological malignancies. Cancer Res. 1997, 5, 837–841. [Google Scholar]
- Batova, A.; Diccianni, M.B.; John, C.Y.; Nobori, T.; Link, M.P.; Pullen, J.; Alice, L.Y. Frequent and selective methylation of p15 and deletion of both p15 and p16 in T-cell acute lymphoblastic leukemia. Cancer Res. 1997, 5, 832–836. [Google Scholar]
- Tsellou, E.; Troungos, C.; Moschovi, M.; Athanasiadou-Piperopoulou, F.; Polychronopoulou, S.; Kosmidis, H.; Kalmanti, M.; Hatzakis, A.; Dessypris, N.; Kalofoutis, A.; et al. Hypermethylation of CpG islands in the promoter region of the p15INK4B gene in childhood acute leukaemia. Eur. J. Cancer 2005, 41, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Matsushita, M.; Zimmermann, M.; Ikezoe, T.; Komatsu, N.; Seriu, T.; Schrappe, M.; Bartram, C.R.; Koeffler, H.P. Clinical significance of aberrant DNA methylation in childhood acute lymphoblastic leukemia. Leuk. Res. 2011, 35, 1345–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mai, H.; Liu, X.; Chen, Y.; Li, C.; Cao, L.; Chen, X.; Chen, S.; Liu, G.; Wen, F. Hypermethylation of p15 gene associated with an inferior poor long-term outcome in childhood acute lymphoblastic leukemia. J. Cancer Res. Clin. Oncol. 2016, 142, 497–504. [Google Scholar] [CrossRef]
- Chim, C.S.; Tam, C.Y.; Liang, R.; Kwong, Y.L. Methylation of p15 and p16 genes in adult acute leukemia: Lack of prognostic significance. Cancer 2001, 12, 2222–2229. [Google Scholar] [CrossRef]
- Hoshino, K.; Asou, N.; Okubo, T.; Suzushima, H.; Kiyokawa, T.; Kawano, F.; Mitsuya, H. The absence of the p15 INK4B gene alterations in adult patients with precursor B-cell acute lymphoblastic leukaemia is a favourable prognostic factor. Br. J. Haematol. 2002, 117, 531–540. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Daniel, J.; Smith, T.L.; Kornblau, S.M.; Lee, M.S.; Kantarjian, H.M.; Issa, J.P.J. DNA methylation of multiple promoter-associated CpG islands in adult acute lymphocytic leukemia. Clin. Cancer Res. 2002, 8, 2217–2224. [Google Scholar]
- Bueso-Ramos, C.; Xu, Y.; McDonnell, T.J.; Brisbay, S.; Pierce, S.; Kantarjian, H.; Rosner, G.; Garcia-Manero, G. Protein Expression of a Triad of Frequently Methylated Genes, p73, p57Kip2, and p15, Has Prognostic Value in Adult Acute Lymphocytic Leukemia Independently of Its Methylation Status. J. Clin. Oncol. 2005, 23, 3932–3939. [Google Scholar]
- Yang, H.; Kadia, T.; Xiao, L.; Bueso-Ramos, C.E.; Hoshino, K.; Thomas, D.A.; O’Brien, S.; Jabbour, E.; Pierce, S.; Rosner, G.L.; et al. Residual DNA methylation at remission is prognostic in adult Philadelphia chromosome–negative acute lymphocytic leukemia. Blood 2009, 113, 1892–1898. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Jeha, S.; Daniel, J.; Williamson, J.; Albitar, M.; Kantarjian, H.M.; Issa, J.P.J. Aberrant DNA methylation in pediatric patients with acute lymphocytic leukemia. Cancer 2003, 97, 695–702. [Google Scholar] [CrossRef]
- Sherborne, A.L.; Hosking, F.J.; Prasad, R.B.; Kumar, R.; Koehler, R.; Vijayakrishnan, J.; Papaemmanuil, E.; Bartram, C.R.; Stanulla, M.; Schrappe, M.; et al. Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat. Genet. 2010, 42, 492–494. [Google Scholar] [CrossRef]
- Hungate, E.A.; Vora, S.R.; Gamazon, E.R.; Moriyama, T.; Best, T.; Hulur, I.; Lee, Y.; Evans, T.J.; Ellinghaus, E.; Stanulla, M.; et al. A variant at 9p21.3 functionally implicates CDKN2B in paediatric B-cell precursor acute lymphoblastic leukaemia aetiology. Nat. Commun. 2016, 7, 1–11. [Google Scholar]
- Xu, H.; Zhang, H.; Yang, W.; Yadav, R.; Morrison, A.C.; Qian, M.; Devidas, M.; Liu, Y.; Perez-Andreu, V.; Zhao, X.; et al. Inherited coding variants at the CDKN2A locus influence susceptibility to acute lymphoblastic leukaemia in children. Nat. Commun. 2015, 6, 1–7. [Google Scholar]
- Walsh, K.M.; De Smith, A.J.; Hansen, H.M.; Smirnov, I.V.; Gonseth, S.; Endicott, A.A.; Xiao, J.; Rice, T.; Fu, C.H.; McCoy, L.S.; et al. A heritable missense polymorphism in CDKN2A confers strong risk of childhood acute lymphoblastic leukemia and is preferentially selected during clonal evolution. Cancer Res. 2015, 75, 4884–4894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudson, A.G. Two genetic hits (more or less) to cancer. Nat. Rev. Cancer 2001, 1, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G. The molecular genetic makeup of acute lymphoblastic leukemia. In Hematology 2010, the American Society of Hematology Education Program Book; American Society of Hematology: Washington, DC, USA, 2012; Volume 2012, pp. 389–396. [Google Scholar]
- Brown, A.L.; De Smith, A.J.; Gant, V.U.; Yang, W.; Scheurer, M.E.; Walsh, K.M.; Chernus, J.M.; Kallsen, N.A.; Peyton, S.A.; Davies, G.E.; et al. Inherited genetic susceptibility to acute lymphoblastic leukemia in down syndrome. Blood 2019, 134, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Toubai, T.; Tanaka, J.; Ota, S.; Fukuhara, T.; Hashino, S.; Kondo, T.; Kasai, M.; Kakinoki, Y.; Masauzi, N.; Morioka, M.; et al. Minimal residual disease (MRD) monitoring using rearrangement of T-cell receptor and immunoglobulin H gene in the treatment of adult acute lymphoblastic leukemia patients. Am. J. Hematol. 2005, 80, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Borowitz, M.J.; Devidas, M.; Hunger, S.P.; Bowman, W.P.; Carroll, A.J.; Carroll, W.L.; Linda, S.; Martin, P.L.; Pullen, D.J.; Viswanatha, D.; et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: A Children’s Oncology Group study. Blood 2008, 111, 5477–5485. [Google Scholar] [CrossRef] [Green Version]
- Stanulla, M.; Dagdan, E.; Zaliova, M.; Bourquin, J.P.; Bornhauser, B. IKZF1 plus Defines a New Minimal Residual Disease–Dependent Very-Poor Prognostic Profile in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2018, 36, 1240–1249. [Google Scholar]
- Den Boer, M.L.; van Slegtenhorst, M.; De Menezes, R.X.; Cheok, M.H.; Buijs-Gladdines, J.G.; Peters, S.T.; Van Zutven, L.J.; Beverloo, H.B.; Van der Spek, P.J.; Escherich, G.; et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: A genome-wide classification study. Lancet Oncol. 2009, 10, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Karrman, K.; Forestier, E.; Heyman, M.; Andersen, M.K.; Autio, K.; Blennow, E.; Borgström, G.; Ehrencrona, H.; Golovleva, I.; Heim, S.; et al. Clinical and cytogenetic features of a population-based consecutive series of 285 pediatric T-cell acute lymphoblastic leukemias: Rare T-cell receptor gene rearrangements are associated with poor outcome. Genes Chromosom. Cancer 2009, 48, 795–805. [Google Scholar] [CrossRef]
- Russo, A.A.; Tong, L.; Lee, J.-O.; Jeffrey, P.D.; Pavletich, N.P. Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature 1998, 395, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, T.; Zindy, F.; Roussel, M.F.; Quelle, D.E.; Downing, J.R.; Ashmun, R.A.; Grosveld, G.; Sherr, C.J. Tumor Suppression at the Mouse INK4a Locus Mediated by the Alternative Reading Frame Product p19 ARF. Cell 1997, 91, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Chin, L.; Pomerantz, J.; Polsky, D.; Jacobson, M.; Cohen, C.; Cordon-Cardo, C.; Horner, J.W.; DePinho, R.A. Cooperative effects of INK4a and ras in melanoma susceptibility in vivo. Genes Dev. 1997, 11, 2822–2834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomerantz, J.; Schreiber-Agus, N.; Liégeois, N.J.; Silverman, A.; Alland, L.; Chin, L.; Potes, J.; Chen, K.; Orlow, I.; Lee, H.W.; et al. The Ink4a tumor suppressor gene product, p19(Arf), interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell 1998, 92, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xiong, Y.; Yarbrough, W.G. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 1998, 92, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Kamijo, T.; Weber, J.D.; Zambetti, G.; Zindy, F.; Roussel, M.F.; Sherr, C.J. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc. Natl. Acad. Sci. USA 1998, 95, 8292–8297. [Google Scholar] [CrossRef] [Green Version]
- Stott, F.J.; Bates, S.; James, M.C.; McConnell, B.B.; Starborg, M.; Brookes, S.; Palmero, I.; Ryan, K.; Hara, E.; Vousden, K.H.; et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998, 17, 5001–5014. [Google Scholar] [CrossRef] [Green Version]
- Serrano, M.; Lee, H.W.; Chin, L.; Cordon-Cardo, C.; Beach, D.; DePinho, R.A. Role of the INK4a locus in tumor suppression and cell mortality. Cell 1996, 85, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Krimpenfort, P.; Quon, K.C.; Mooi, W.J.; Loonstra, A.; Berns, A. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature 2001, 413, 83–86. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Bardeesy, N.; Lee, K.H.; Carrasco, D.; Castrillon, D.H.; Aguirre, A.J.; Wu, E.A.; Horner, J.W.; DePinho, R.A. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001, 413, 86–91. [Google Scholar]
- Williams, R.T.; Sherr, C.J. The INK4-ARF (CDKN2A/B) locus in hematopoiesis and BCR-ABL-induced leukemias. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 461–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, J.L.; Kieboom, K.; Marino, S.; DePinho, R.A.; Van Lohuizen, M. The oncogene and Polycombgroup gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999, 397, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Lessard, J.; Sauvageau, G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 2003, 423, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Park, I.K.; Qian, D.; Kiel, M.; Becker, M.W.; Pihalja, M.; Weissman, I.L.; Morrison, S.J.; Clarke, M.F. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 2003, 423, 302–305. [Google Scholar] [CrossRef]
- Iwama, A.; Oguro, H.; Negishi, M.; Kato, Y.; Morita, Y.; Tsukui, H.; Ema, H.; Kamijo, T.; Katoh-Fukui, Y.; Koseki, H.; et al. Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity 2004, 21, 843–851. [Google Scholar] [CrossRef] [Green Version]
- Akala, O.O.; Park, I.K.; Qian, D.; Pihalja, M.; Becker, M.W.; Clarke, M.F. Long-term haematopoietic reconstitution by Trp53-/-p16 Ink4a-/-p19Arf-/- multipotent progenitors. Nature 2008, 453, 228–232. [Google Scholar] [CrossRef]
- Volanakis, E.J.; Boothby, M.R.; Sherr, C.J. Epigenetic regulation of the Ink4a-Arf (Cdkn2a) tumor suppressor locus in the initiation and progression of Notch1-driven T cell acute lymphoblastic leukemia. Exp. Hematol. 2013, 41, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Volanakis, E.J.; Williams, R.T.; Sherr, C.J. Stage-specific Arf tumor suppression in Notch1-induced T-cell acute lymphoblastic leukemia. Blood 2009, 114, 4451–4459. [Google Scholar] [CrossRef] [Green Version]
- Chan, L.C.; Karhi, K.K.; Rayter, S.I.; Heisterkamp, N.; Eridani, S.; Powle, R.; Lawler, S.D.; Groffen, J.; Foulkes, J.G.; Greaves, M.F.; et al. A novel abl protein expressed in Philadelphia chromosome positive acute lymphoblastic leukaemia positive acute lymphoblastic leukaemia. Nature 1987, 325, 635–637. [Google Scholar] [CrossRef]
- Clark, S.S.; Mclaughlin, J.; Crist, W.M.; Champlin, R.; Witte, O.N. Unique forms of the abl tyrosine kinase distinguish Ph1-positive CML from Ph1-positive ALL. Science 1987, 235, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Castor, A.; Nilsson, L.; Åstrand-Grundström, I.; Buitenhuis, M.; Ramirez, C.; Anderson, K.; Strömbeck, B.; Garwicz, S.; Békássy, A.N.; Schmiegelow, K.; et al. Distinct patterns of hematopoietic stem cell involvement in acute lymphoblastic leukemia. Nat. Med. 2005, 11, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Signer, R.A.J.; Montecino-Rodriguez, E.; Witte, O.N.; Dorshkind, K. Immature B-cell progenitors survive oncogenic stress and efficiently initiate Ph+ B-acute lymphoblastic leukemia. Blood 2010, 116, 2522–2530. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.T.; Roussel, M.F.; Sherr, C.J. Arf gene loss enhances oncogenicity and limits imatinib response in mouse models of Bcr-Abl-induced acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2006, 103, 6688–6693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.Y.; Young, F.; Chen, C.Y.; Stevens, B.M.; Neering, S.J.; Rossi, R.M.; Bushnell, T.; Kuzin, I.; Heinrich, D.; Bottaro, A.; et al. The biologic properties of leukemias arising from BCR/ABL-mediated transformation vary as a function of developmental origin and activity of the p19ARF gene. Blood 2008, 112, 4184–4192. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Williams, R.T.; Downing, J.R.; Sherr, C.J. Failure of CDKN2A/B (INK4A/B-ARF)-mediated tumor suppression and resistance to targeted therapy in acute lymphoblastic leukemia induced by BCR-ABL. Genes Dev. 2008, 22, 1411–1415. [Google Scholar] [CrossRef] [Green Version]
- Treanor, L.M.; Volanakis, E.J.; Zhou, S.; Lu, T.; Sherr, C.J.; Sorrentino, B.P. Functional interactions between Lmo2, the Arf tumor suppressor, and Notch1 in murine T-cell malignancies. Blood 2011, 117, 5453–5462. [Google Scholar] [CrossRef] [Green Version]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Vassiliou, L.V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; DiTullio, R.A.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef]
- Bartkova, J.; Hořejší, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Denko, N.C.; Giaccia, A.J.; Stringer, J.R.; Stambrook, P.J. The human Ha-ras oncogene induces genomic instability in murine fibroblasts within one cell cycle. Proc. Natl. Acad. Sci. USA 1994, 91, 5124–5128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spruck, C.H.; Won, K.-A.; Reed, S.I. Deregulated cyclin E induces chromosome instability. Nature 1999, 401, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S. Deregulated G1-cyclin expression induces genomic instability by preventing efficient pre-RC formation. Genes Dev. 2002, 16, 2639–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsantoulis, P.K.; Kotsinas, A.; Sfikakis, P.P.; Evangelou, K.; Sideridou, M.; Levy, B.; Mo, L.; Kittas, C.; Wu, X.R.; Papavassiliou, A.G.; et al. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene 2008, 27, 3256–3264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, A.J.; Stampfer, M.R.; Aldaz, C.M. Increased p16 expression with first senescence arrest in human mammary epithelial cells and extended growth capacity with p16 inactivation. Oncogene 1998, 17, 199–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Counter, C.M.; Avilion, A.A.; LeFeuvre, C.E.; Stewart, N.G.; Greider, C.W.; Harley, C.B.; Bacchetti, S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992, 5, 1921–1929. [Google Scholar] [CrossRef]
- Rogan, E.M.; Bryan, T.M.; Hukku, B.; Maclean, K.; Chang, A.C.; Moy, E.L.; Englezou, A.; Warneford, S.G.; Dalla-Pozza, L.; Reddel, R.R. Alterations in p53 and p16INK4 expression and telomere length during spontaneous immortalization of Li-Fraumeni syndrome fibroblasts. Mol. Cell. Biol. 1995, 15, 4745–4753. [Google Scholar] [CrossRef] [Green Version]
- De Lange, T.; Shiue, L.; Myers, R.M.; Cox, D.R.; Naylor, S.L.; Killery, A.M.; Varmus, H.E. Structure and variability of human chromosome ends. Mol. Cell. Biol. 1990, 10, 518–527. [Google Scholar] [CrossRef] [Green Version]
- Hastie, N.D.; Dempster, M.; Dunlop, M.G.; Thompson, A.M.; Green, D.K.; Allshire, R.C. Telomere reduction in human colorectal carcinoma and with ageing. Nature 1990, 346, 866–868. [Google Scholar] [CrossRef]
- Harley, C.B.; Vaziri, H.; Counter, C.M.; Allsopp, R.C. The telomere hypothesis of cellular aging. Exp. Gerontol. 1992, 27, 375–382. [Google Scholar] [CrossRef]
- Radpour, R.; Barekati, Z.; Haghighi, M.M.; Kohler, C.; Asadollahi, R.; Torbati, P.M.; Holzgreve, W.; Zhong, X.Y. Correlation of telomere length shortening with promoter methylation profile of p16/Rb and p53/p21 pathways in breast cancer. Mod. Pathol. 2010, 23, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Britigan, E.M.C.; Wan, J.; Zasadil, L.M.; Ryan, S.D.; Weaver, B.A. The ARF tumor suppressor prevents chromosomal instability and ensures mitotic checkpoint fidelity through regulation of Aurora, B. Mol. Biol. Cell 2014, 25, 2761–2773. [Google Scholar] [CrossRef] [PubMed]
- Healy, J.; Bélanger, H.; Beaulieu, P.; Larivière, M.; Labuda, D.; Sinnett, D. Promoter SNPs in G1/S checkpoint regulators and their impact on the susceptibility to childhood leukemia. Blood 2007, 109, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Maloney, K.W.; McGavran, L.; Odom, L.F.; Hunger, S.P. Acquisition of p16(INK4A) and p15(INK4B) gene abnormalities between initial diagnosis and relapse in children with acute lymphoblastic leukemia. Blood 1999, 93, 2380–2385. [Google Scholar] [CrossRef] [PubMed]
- Carter, T.L.; Reaman, G.H.; Kees, U.R. INK4A/ARF deletions are acquired at relapse in childhood acute lymphoblastic leukaemia: A paired study on 25 patients using real-time polymerase chain reaction. Br. J. Haematol. 2001, 113, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Davidsson, J.; Paulsson, K.; Lindgren, D.; Lilljebjörn, H.; Chaplin, T.; Forestier, E.; Andersen, M.K.; Nordgren, A.; Rosenquist, R.; Fioretos, T.; et al. Relapsed childhood high hyperdiploid acute lymphoblastic leukemia: Presence of preleukemic ancestral clones and the secondary nature of microdeletions and RTK-RAS mutations. Leukemia 2010, 24, 924–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Waanders, E.; van Reijmersdal, S.V.; Antić, Ž.; van Bosbeek, C.M.; Sonneveld, E.; de Groot, H.; Fiocco, M.; van Kessel, A.G.; van Leeuwen, F.N.; et al. Upfront Treatment Influences the Composition of Genetic Alterations in Relapsed Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. HemaSphere 2020, 4. [Google Scholar] [CrossRef]
- Soenen, V.; Lepelley, P.; Gyan, E.; Preudhomme, C.; Lai, J.L.; Bauters, F.; Fenaux, P.; Quesnel, B. Prognostic significance of p16INK4a immunocytochemistry in adult ALL with standard risk karyotype. Leukemia 2001, 15, 1054–1059. [Google Scholar] [CrossRef] [Green Version]
- Kuster, L.; Grausenburger, R.; Fuka, G.; Kaindl, U.; Krapf, G.; Inthal, A.; Mann, G.; Kauer, M.; Rainer, J.; Kofler, R.; et al. ETV6/RUNX1-positive relapses evolve from an ancestral clone and frequently acquire deletions of genes implicated in glucocorticoid signaling. Blood 2011, 117, 2658–2667. [Google Scholar] [CrossRef]
- Spinella, J.-F.; Richer, C.; Cassart, P.; Ouimet, M.; Healy, J.; Sinnett, D. Mutational dynamics of early and late relapsed childhood ALL: Rapid clonal expansion and long-term dormancy. Blood Adv. 2018, 2, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, M.P.; Bastian, L.; Eckert, C.; Gökbuget, N.; James, A.R.; Tanchez, J.O.; Schlee, C.; Isaakidis, K.; Häupl, B.; Baum, K.; et al. Integrated analysis of relapsed B-cell precursor Acute Lymphoblastic Leukemia identifies subtype-specific cytokine and metabolic signatures. Sci. Rep. 2019, 9, 1–11. [Google Scholar]
- Williams, R.T.; Den Besten, W.; Sherr, C.J. Cytokine-dependent imatinib resistance in mouse BCR-ABL+, Arf-null lymphoblastic leukemia. Genes Dev. 2007, 21, 2283–2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawai, C.M.; Freund, J.; Oh, P.; Ndiaye-Lobry, D.; Bretz, J.C.; Strikoudis, A.; Genesca, L.; Trimarchi, T.; Kelliher, M.A.; Clark, M.; et al. Therapeutic Targeting of the Cyclin D3:CDK4/6 Complex in T Cell Leukemia. Cancer Cell 2012, 22, 452–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheppard, K.E.; McArthur, G.A. The cell-cycle regulator CDK4: An emerging therapeutic target in melanoma. Clin. Cancer Res. 2013, 19, 5320–5328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, M.A. Molecular pathways: CDK4 inhibitors for cancer therapy. Clin. Cancer Res. 2014, 20, 3379–3383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, J.L.; Thangavel, C.; McClendon, A.K.; Reed, C.A.; Knudsen, E.S. Therapeutic CDK4/6 inhibition in breast cancer: Key mechanisms of response and failure. Oncogene 2010, 29, 4018–4032. [Google Scholar] [CrossRef] [Green Version]
- Raub, T.J.; Wishart, G.N.; Kulanthaivel, P.; Staton, B.A.; Ajamie, R.T.; Sawada, G.A.; Gelbert, L.M.; Shannon, H.E.; Sanchez-Martinez, C.; De Dios, A. Exposure of Two Selective Dual CDK4 and CDK6 Inhibitors and the Antitumor Activity of CDK4 and CDK6 Inhibition in Combination with Temozolomide in an Intracranial Glioblastoma Xenograft. Drug Metab. Dispos. 2015, 43, 1360–1371. [Google Scholar] [CrossRef] [Green Version]
- Fry, D.W.; Harvey, P.J.; Keller, P.R.; Elliott, W.L.; Meade, M.; Trachet, E.; Albassam, M.; Zheng, X.; Leopold, W.R.; Pryer, N.K.; et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 2004, 3, 1427–1438. [Google Scholar]
- Kim, S.; Tiedt, R.; Loo, A.; Horn, T.; Delach, S.; Kovats, S.; Haas, K.; Engstler, B.S.; Cao, A.; Pinzon-Ortiz, M.; et al. The potent and selective cyclin-dependent kinases 4 and 6 inhibitor ribociclib (LEE011) is a versatile combination partner in preclinical cancer models. Oncotarget 2018, 9, 35226. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Reference | Trial or Patient Origin (Period) | Cohort Size | Age (y) | Type of ALL | Frequency del (Method) | EFS/DFS/RFS (p) | OS (p) | CIR (p) |
|---|---|---|---|---|---|---|---|---|
| [36] | CCG-1881, 1882, 1891, 1922 (1988 to 1995) | 864 | 1–18 | BCP | 9p abn. 12% (Karyotyping) | Univariate: EFS 6y-9p abn 63% vs. no 9p abn 77% (p = 0.0004) | - | - |
| [37] | (1987–1997) | 194 | 1–15 | BCP | CDKN2A del homo 24%, ARF del homo 27%, CDKN2B del homo 18% (Southern blot, SSCP, Sanger sequencing) | Univariate: EFS CDKN2A del homo 0.58 vs. 0.77 (p < 0.001) Multivariate: CDKN2A del homo poor (p < 0.01) | - | - |
| [38] | DCOG ALL8 and 9 (1991–2004) | 109 | 0–17 | BCP | CDKN2A/B del 34% (FISH) | - | Univariate: 4y-CDKN2A/B del 80% vs. 87% (p = ns) Multivariate: CDKN2A/B del HR = 1.254 (p = 0.652) | Univariate: 4y-CDKN2A/B del 73% vs. 74% (p = ns) Multivariate: CDKN2A/B del HR = 1.251 (p = 0.608) |
| [39] | EORTC 58881 and 58951 (1989–2001) | 227 | 0–17 | BCP | CDKN2A del 31%, CDKN2B del 23% (qPCR) | Univariate: 6yEFS-CDKN2A del homo 68% vs. CDKN2A del hetero 80% vs. CDKN2A wt 75% (p = ns) | Univariate: CDKN2A del homo 82% vs. CDKN2A del hetero 90% vs. wt 87% (p = ns) | Univariate: CDKN2A del homo 11 vs. CDKN2A del hetero 5 vs. wt 32 (p = ns) |
| [40] | Disc. COG P9906 (2000–2003) Val. multiple COG protocols (1986–2007) | 479 | <18 | Disc: 221 high-risk BCP Val: 258 BCP | CDKN2A/B del 46% disc. CDKN2A/B del 38% val. (SNPa) | Univariate: ns (outcome data not shown) | Univariate: ns (outcome data not shown) | Univariate: ns (outcome data not shown) |
| [41] | NOPHO2000 (2002–2006) | 452 | 1–14 | BCP | CDKN2A/B del 16% (FISH) | Univariate: 5yEFS-CDKN2A/B del homo 76% vs. CDKN2A/B del hetero 76% vs. CDKN2A/B wt 83% (p = 0.330) | - | - |
| [42] | ALL-REZ BFM 2002 (2002–2009) | 294 | 0–18 | BCP at 1st relapse | CDKN2A/B del 37% (MLPA) | Univariate: EFS CDKN2A/B del 0.45 vs. wt 0.43 (p = 0.990) | Univariate: CDKN2A/B del 0.48 vs. wt 0.54 (p = 0.443) | Univariate: CDKN2A/B del 0.40 vs. wt 0.21 (p = 0.001) |
| [43] | PETHEMA (1996–2014) | 115 | 0–17 | BCP | CDKN2A/B del 33% (CGH array) | Univariate: EFS ns (outcome data not shown) | Univariate: ns (outcome data not shown) | Univariate: ns (outcome data not shown) |
| [19] | ALL IC BFM 2002 and 2009 (2002–-2017) | 641 | 2–12 | BCP | CDKN2A del 26%, CDKN2B del 22% (MLPA, SNPa) | Univariate: RFS CDKN2A del homo HR 2.21 (p = 0.028) Multivariate: CDKN2A del homo HR = 3.09 (p = 0.007) | Univariate: 2y-CDKN2A/B del 85% vs. wt 88% (p = 0.560) | - |
| [44] | GIMEMA 2000-0904-1104-1308 and AIEOP ALL 2000, AIEOP-BFM ALL 2009 (2000–2018) | 157 | 1–15 (n = 45) | BCP negative for BCR-ABL1, ETV6-RUNX1, TCF3-PBX1 or KMT2Ar | CDKN2A/B del 11% (MLPA) | Multivariate (children + AYA + adults): CDKN2A/B/RB1 HR = 2.12 (p = 0.048) | Univariate: ns (outcome data not shown) | Univariate: ns (outcome data not shown) |
| [45] | ANZCHOG ALL8 (2002–2011) | 475 | 1–18 | Non-high-risk BCP | CDKN2A/B del 36% (MLPA) | Univariate: 7y-EFS CDKN2A/B del homo 77% vs. del hetero 81% vs. wt 80% (p = ns) | Univariate: 7y-CDKN2A/B del homo 87% vs. del hetero 93% vs. wt 94% (p < 0.05) | Univariate: 7y-CDKN2A/B del homo 18% vs. del hetero 17% vs. wt 17% (p = ns) |
| [27] | DCOG-ALL10 (2004–2012) | 515 | 1–18 | BC | CDKN2A/B del 33% (MLPA) | Univariate: EFS CDKN2A/B del 79% vs. wt 87% (p = ns) | Univariate: ns (outcome data not shown) | Univariate: CDKN2A/B del 17% vs. wt 10% (p = ns) |
| [46] | ALLR3 (2003–2013) | 192 | 1–18 | 1st (late) relapse BCP | CDKN2A/B del 22% (MLPA) | Univariate: 5y-CDKN2A/B del 63% vs. wt 62% (p = 0.75) | Univariate: 5y-CDKN2A/B del 69% vs. wt 75% (p = 0.26) | |
| [35] | ICICLE (Indian adaption of UKMRC2007 protocol) (2015–2017) | 83 | 1–12 | BCP | DKN2A/B del 36% (MLPA) | Univariate: 28month-EFS CDKN2A/B del 42% vs. wt 90% (p = 0.0004) Multivariate: CDKN2A/B del HR = 5.75 (p = 0.008) | - | - |
| [47] | St Jude Children’s Research Hospital (1993–2005) | 50 | <18 | T-ALL | CDKN2A/B del 72% (SNP array) | - | - | - |
| [24] | UKALLXI ALL97-2003 (1986–2007) | 266 | <18 | T-ALL | CDKN2A/B del 50% (SNP array, CGHa, FISH) | - | - | - |
| [48] | St Jude, the Children’s Oncology Group and AIEOP | ETP 42 Non-ETP 64 | <18 | T-ALL | ETP: CDKN2A del 25% Non-ETP: CDKN2A del 81% (SNPa) | - | Univariate. 5y-CDKN2A del 24.2% vs. wt 35.8% (p = 0.2814) | |
| [49] | NOPHO ALL-1981–1986–1992–2000–2008 (1983–2011) | 47 | 0-18 | T-ALL | CDKN2A del 72% CDKN2B del 62.5% (SNPa) | Univariate: 5y-EFS CDKN2A del 0.48 vs. wt 0.73 (p = ns) | Univariate: 5y-CDKN2A del 0.52 vs. wt 0.91 (p = 0.04) | - |
| [50] | France and UK | 155 | 111 c. 44 a. | T-ALL | CDKN2A del 78% (FISH, MLPA, CGHa, TDS) | - | - | - |
| [43] | PETHEMA (1996–2014) | 27 | <18 | T-ALL | CDKN2A/B del 70.4% (CGHa) | Univariate: ns (outcome data not shown) | Univariate: ns (outcome data not shown) | Univariate: ns (outcome data not shown) |
| [51] | Children’s Oncology Group trial AALL0434 (2007–2011) | 264 | 1–29 | T-ALL | CDKN2A/B del 78.4% (SNPa) | Univariate: 5yEFS-CDKN2A del 90.6% vs. wt 92.7% (p = 0.349) | Univariate: 5y-CDKN2A del 94.5% vs. wt 100% (p = 0.0466) | Univariate: 5y-CDKN2A del 7.9% vs. wt 7.2% (p = 0.6953) |
| [52] | TPOG-ALL-93 (1995–2015) | 102 | <18 | T-ALL | CDKN2A del 63.3%, CDKN2B del 50% (MLPA) | - | Univariate: ns (outcome data not shown) | Univariate: ns (outcome data not shown) |
| [53] | Brazilian Group Childhood Leukemia 99 (2005–2017) | 341 | <19 | T-ALL | CDKN2A/B del 71.4% (MLPA) | - | Univariate: 5y-CDKN2A/B del 62.6% vs. wt 62.5% (p = 0.729) | - |
| [54] | Indian Childhood Collaborative Leukemia (ICICLE) (2017–2018) | 27 | <18 | T-ALL | CDKN2A/B del 59.2% (digital MLPA) | - | Univariate: ns (outcome data not shown) | - |
| Reference | Trial or Patient Origin (Period) | Cohort Size | Age (y) | Type of ALL | Frequency del (Method) | EFS/DFS/RFS (p) | OS (p) | CIR (p) |
|---|---|---|---|---|---|---|---|---|
| [55] | MRC UKALLXII/ECOG E2993 (1993–2004) | 796 | 15–65 | Ph− BCP | del(9p) 9% (Karyotyping) | Univariate: 5y-EFS del(9p) 49%, O/E 0.73 (p = 0.043) | Univariate: 5y-del(9p) 58%, O/R 0.70 (p = 0.032) | - |
| [56] | L-10 and Swedish ALL group protocol (1986–2006) | 240 | 17–78 | BCP | 9p abn. 7% (Karyotyping) | Univariate: median EFS 9p abn 6 months vs. no 9p abn 2.5 years, (p = 0.0134) | Univariate: median OS 9p abn 5 months vs. no 9p abn+ no HSCT 5y (p = 0.023) Multivariate: 9p abn RR = 2.21 (p = 0.032) | - |
| [57] | Japan Adult Leukemia Study Group (JALSG) (2002–2005) | 80 | 15–64 | Ph+ BCP | 9p abn. 10% (Karyotyping) | Univariate: lower RFS, (p = 0.005) | - | - |
| [18] | GIMEMA LAL0201-2000 and LAL1205 (1996–2008) | 101 | 18–76 | Ph+ BCP | CDKN2A del 29%, CDKN2B del 25% (SNPa, FISH) | Univariate: 2y-DFS CDKN2A/B del 22% vs. wt 58% (p = 0.001) Multivariate: CDKN2A/B del poor DFS (p = 0.005) | Univariate: 2y-CDKN2A/B del 57% vs. wt 78% (p = 0.02) | Univariate: 2y-CDKN2A/B del 73% vs. wt 38% (p = 0.001) |
| [58] | UKALLXII/ECOG2993 (1993–2006) | 454 | 15–65 | Ph− BCP | CDKN2A/B del 24% (MLPA, FISH) | Univariate: 5y-EFS CDKN2A/B del 39% HR = 1.20 (p = 0.247) 5y-EFS CDKN2A/B homo del vs. mono del HR = 0.59 (p = 0.08) | Univariate: 5y-CDKN2A/B del 42%, HR= 1.16 (p = 0.366) | - |
| [59] | PETHEMA AR93-03, OLD07, RI96-RI08 and Ph08 (1993–2013) | 152 | 15–74 | BCP | CDKN2A/B del 42% (MLPA) | - | Univariate: 5y-CDKN2A/B del 25% vs. wt 57% (p = 0.001); 5y-Ph+ CDKN2A/B del 14% vs. 54% (p = 0.025) Multivariate: CDKN2A/B del HR = 2.545 (p < 0.001) | Univariate: CDKN2A/B del 54% vs. wt 41% (p = 0.063); 5y-Ph+ CDKN2A/B del 100% vs. 43% (p = 0.071) |
| [60] | Chinese Han-South Medical University (2008–2013) | 215 | 15–60 | BCP | Diagnosis: CDKN2A/B del 28% 1st relapse: CDKN2A/B del 45% (FISH) | Univariate diagnosis: EFS CDKN2A/B del 12 vs. wt 24 months (p < 0.0001) Univariate 1st relapse: EFS CDKN2A/B del 5 vs. wt 16 months (p = 0.004) | Univariate diagnosis: CDKN2A/B del 19 vs. wt 30 months (p < 0.0001) Univariate 1st relapse: CDKN2A/B del 8 vs. wt 18 months (p = 0.001) | Univariate diagnosis: 2y-CDKN2A/B del 59% vs. wt 36% (p = 0.002) |
| [43] | PETHEMA AR93-03-11, RI96, OLD07, Ph00-08 (1996–2014) | 100 | 18–84 | BCP | CDKN2A/B del 47% (CGHa) | Univariate: ns (outcome data not shown) | Univariate: ns (outcome data not shown) | Univariate: ns (outcome data not shown) |
| [61] | Chinese Han-South Medical University (2008–2014) | 135 | 18–65 | Ph+ BCP | CDKN2A/B del 33% (FISH) | Univariate: 2y-DFS CDKN2A/B del 23% vs. wt 35% (p = 0.005) | Univariate: 2y-CDKN2A/B del 51% vs. wt 65% (p = 0.004) | Univariate: 2y-CDKN2A/B del 59% vs. wt 35% (p = 0.008) |
| [62] | Asan Medical Center, Korea. (2000–2015) | 122 | 19–74 | Ph+ BCP | del(9p) 20% (Karyotyping) | Univariate: 5y-DFS del(9p) 34% vs. wt 61% ( p= 0.189) Multivariate: DFS del(9p) HR = 3.42 (p = 0.002) | Univariate: 5y-del(9p) 44% vs. wt 76% (p = 0.091) Multivariate: del(9p) HR = 2.16 (p = 0.031) | - |
| [63] | Huntsman Cancer Institute (UT) and, Ann Arbor (MI) and Intermountain Healthcare (UT) (1998–2016) | 70 | 18-83 | BCP | CDKN2A/B del 49% (SNPa) | Univariate: median EFS CDKN2A/B del 9.5 months HR = 1.10 (p = ns) | Univariate: median OS CDKN2A/B del 21.8 months HR = 1.36 (p = ns); CDKN2A/B + IKZF1 del HR = 2.6 (p = 0.0007) | - |
| [64] | MD Anderson cohort (2001–2016) | 182 | 19–85 | Ph+ BCP | del(9p)-16% (Karyotyping) | Univariate: 5-y RFS del(9p) 34% (p = ns) | Univariate: 5y-del(9p) 26% (p = ns) | - |
| [65] | GRAALL 2003–2005 (2003–2011) | 542 | 15–59 | Ph− BCP | del(9p) 12% (Karyotyping) | Univariate: EFS del(9p) HR = 1.05 (p = 0.78) | Univariate: del(9p) HR = 0.86 (p = 0.46) | Univariate: del(9p) SHR = 1.10 (p = 0.65) |
| [44] | GIMEMA 2000-0904-1104-1308 and AIEOP LLA 2000, AIEOP-BFM ALL 2009 (2000–2018) | 157 | 15–35 (n = 56) 36–78 (n = 56) | BCP negative for BCR-ABL1, ETV6-RUNX1, TCF3-PBX1 or KMT2Ar | 15–35 CDKN2A/B del: 48% 36–78 CDKN2A/B del: 46% (MLPA) | Univariate: 5y-DFS A. CDKN2A/B and/or RB1 del 13% vs. wt 54% (p = 0.03) Multivariate: (all ages): CDKN2A/B/RB1 del HR = 2.12 (p = 0.048) | Univariate: ns (outcome data not shown) | Univariate: ns (outcome data not shown) |
| [26] | GMALL 06/99 and 07/2003 (2001–2009) | 97 | 18–64 | Ph+ BCP | CDKN2A/B del 41% (SNPa, MLPA) | Multivariate: DFS CDKN2A/B del HR 2.621 (p = 0.0054) | Multivariate: CDKN2A/B del HR 2.162 (p = 0.014) | - |
| [66] | GIMEMA LAL0201B-0904-1205-1509 (2000–2018) | 116 | 18–89 | Ph+ BCP | CDKN2A/B del 32% (SNPa, MLPA) | 3y-DFS IKZF1 + CDKN2A/B and/or PAX5 del 25% vs. 43% IKZF1 del only (p = 0.026) Multivariate: DFS CDKN2A/B del HR = 1.608 (p = 0.089) | 3y-IKZF1 + CDKN2A/B and/or PAX5 del 40% vs. 63% IKZF1 del only (p = 0.02) | Univariate: ns (outcome data not shown) |
| [67] | PETHEMA AR93-03-11, OLD07, RI96-08 (1993–2017) | 128 | 15–75 | Ph− BCP | CDKN2A/B del 44% (MLPA) | Univariate: 5-y DFS CDKN2A/B del 25% vs. wt 47% (p = 0.027) | Univariate: 5y-CDKN2A/B del 34% vs. wt 57% (p = 0.042) Multivariate: CDKN2A/B del HR = 2.216 (p = 0.023) | Univariate: 5-y CDKN2A/B del 56% vs. wt 41% (p = 0.090) |
| [68] | PETHEMA AR03 and AR11 (2003–2017) | 44 | 16–59 | BCP negative for BCR-ABL1, ETV6-RUNX1, TCF3-PBX1, KMT2Ar, high hyperdiploid and low hypodiploid | CDKN2A/B del 43% (MLPA) | Univariate: DFS CDKN2A/B del HR = 2.861 (p = 0.032) Multivariate: DFS CDKN2A/B del HR = 2.940 (p = 0.064) | Univariate: CDKN2A/B del HR = 2.523 (p = 0.073) Multivariate: CDKN2A/B del HR = 4.039 (p = 0.029) | Univariate: CIR CDKN2AB del HR = 2.900 (p = 0.039) |
| [69] | UKALL XII/ECOG 2993 (1993–2006) | 108 | >18 | T-ALL | CDKN2A/B del 42% (FISH) | - | Univariate: 5y-CDKN2A del 52% (33;71) | - |
| [70] | GMALL 07/2003 and GMALL Elderly 01/2003 | 90 | 18–88 | T-ALL | CDKN2A/B del 43% (FISH) | - | Univariate: 2y-CDKN2A/B del 77.2% vs. wt 47.2% (p = 0.076) | - |
| [71] | UKALL XII/ECOG 2993 | 53 | >18 | T-ALL | CDKN2A/B del 41% (CGHa) | - | Univariate: 5y-CDKN2A/B del homo 71% vs. del hetero 38% (p = 0.0119) | - |
| [72] | Lithuania (2007–2013) | 25 | 18–64 | T-ALL | CDKN2A/B del 28% (SNPa) | Univariate: ns (outcome data not shown) | Univariate: ns (outcome data not shown) | - |
| [43] | PETHEMA AR93-03, AR11, RI96, OLD07, Ph00-08 (1996–2014) | 23 | 18–84 | T-ALL | CDKN2A/B del 8.7% (CGHa) | Univariate: ns (outcome data not shown) | Univariate: ns (outcome data not shown) | Univariate: ns (outcome data not shown) |
| [73] | Institute of Hematology and Blood Diseases Hospital (China) (2009–2015) | 18 | 14–61 | T-ALL | CDKN2A del 50% CDKN2B del 33.3% (MLPA) | Univariate: ns (outcome data not shown) | Univariate: ns (outcome data not shown) | Univariate: ns (outcome data not shown) |
| [74] | PETHEMA HR-2003-11 (2003–2017) | 62 | 16–72 | T-ALL | CDKN2A del 50% CDKN2B del 47% (qPCR) | - | Univariate: 3y- CDKN2A/B del 75% vs. wt 36% (p = 0.05) | - |
| [75] | Seoul St. Mary’s Hospital (2004–2015) | 102 | 2–77 | T-ALL | CDKN2A/B del 45.1% (MLPA) | - | Univariate: ns (outcome data not shown) | - |
| Reference | Type of ALL | Cohort Size | Age (y) | Technique | Frequency of Methylation | Prognosis | ||
|---|---|---|---|---|---|---|---|---|
| CDKN2A (n) | CDKN2B (n) | CDKN2A | CDKN2B | |||||
| [89] | BCP | 23 | <18 | MS-PCR | 0% (23) | 48% (23) | - | |
| T-ALL | 12 | 0% (12) | 50% (12) | - | ||||
| [90] | T-ALL | 45 | <18 | MS-PCR | 11.7% (17) | 68% (25) | - | |
| [91] | BCP | 36 | <18 | MS-PCR | - | 13% (23) | - | |
| T-ALL | 46.2 (13) | |||||||
| [39] | BCP | 227 | 0–17 | MS-PCR | 13% (31) | 37.5% (28) | Non-significant | |
| [92] | BCP and T-ALL | 95 | <18 | MS-PCR | 4% (95) | 25% (95) * | Non-significant | |
| [19] | BCP | 333 | <18 | MS-MLPA | 3.9% (333) | 87% (333) | Non-significant | Univariate: trend to poor OS |
| [93] | BCP | 93 | 1–13 | MS-PCR | - | 57% (21) | - | Univariate: EFS-8y hyper.71% vs. hypo 91% (p = 0.02); rate of relapse hyper 28% vs. hypo 9.3% (p = 0.02) |
| T-ALL | - | 38% (72) | ||||||
| Reference | Type of ALL | Cohort Size | Age (y) | Technique | Frequency of Methylation | Prognosis | ||
|---|---|---|---|---|---|---|---|---|
| CDKN2A (n) | CDKN2B (n) | CDKN2A | CDKN2B | |||||
| [94] | BCP | 41 | >18 | MS-PCR | 12.5% (41) | 2.4% (41) | - | Univariate: 5y-OS methy 12% vs. un-methy 36% (p = 0.84); 5y-DFS methy 7% vs. un-methy 19% (p = 0.98) |
| T-ALL | 8 | 62.5% (8) | 39% (8) | |||||
| [95] | BCP | 70 | >18 | MS-PCR | 23% (70) | 37% (70) | - | Multivariate: normal CDKN2B was a favourable prognostic factor for longer DFS (p = 0.0001) |
| [96] | BCP | 80 | >18 | MS-PCR | 2.5% (80) | 22.5% (71) | Univariate: Ph− (n = 57), 5y-OS methy 50% vs. un-methy 42% (p = 0.8) | Univariate: Ph− (n = 57) 5y-OS methy 26% vs. un-methy 46% (p = 0.09) |
| T-ALL | Non-significant | Non-significant | ||||||
| [97] | BCP and T-ALL | 64 | 16–78 | MS-PCR | - | 25% (64) | - | Non-significant |
| [98] | Ph− and MLL-BCP | 199 | 15–83 | Real Time bisulfite PCR | - | 17.4% (189) | - | Non-significant |
| [70] | T-ALL | 90 | >18 | MS-PCR | - | 48.6% (74) * | - | - |
| [75] | T-ALL | 102 | 2–77 | pyrosequencing | 3.8% (93) | 50.6% (93) ** | - | Univariate: 3y-EFS high methy 35.9% vs. low methy 59.1% (p = 0.042) Multivariate: CDKN2B biallelic deletion or high methylation HR = 6.358 (p = 0.012) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Gil, C.; Ribera, J.; Ribera, J.M.; Genescà, E. The Yin and Yang-Like Clinical Implications of the CDKN2A/ARF/CDKN2B Gene Cluster in Acute Lymphoblastic Leukemia. Genes 2021, 12, 79. https://doi.org/10.3390/genes12010079
González-Gil C, Ribera J, Ribera JM, Genescà E. The Yin and Yang-Like Clinical Implications of the CDKN2A/ARF/CDKN2B Gene Cluster in Acute Lymphoblastic Leukemia. Genes. 2021; 12(1):79. https://doi.org/10.3390/genes12010079
Chicago/Turabian StyleGonzález-Gil, Celia, Jordi Ribera, Josep Maria Ribera, and Eulàlia Genescà. 2021. "The Yin and Yang-Like Clinical Implications of the CDKN2A/ARF/CDKN2B Gene Cluster in Acute Lymphoblastic Leukemia" Genes 12, no. 1: 79. https://doi.org/10.3390/genes12010079
APA StyleGonzález-Gil, C., Ribera, J., Ribera, J. M., & Genescà, E. (2021). The Yin and Yang-Like Clinical Implications of the CDKN2A/ARF/CDKN2B Gene Cluster in Acute Lymphoblastic Leukemia. Genes, 12(1), 79. https://doi.org/10.3390/genes12010079