Non-Redundant tRNA Reference Sequences for Deep Sequencing Analysis of tRNA Abundance and Epitranscriptomic RNA Modifications




Abstract
:1. Introduction
2. Materials and Methods
2.1. Library Preparations
2.2. Computations
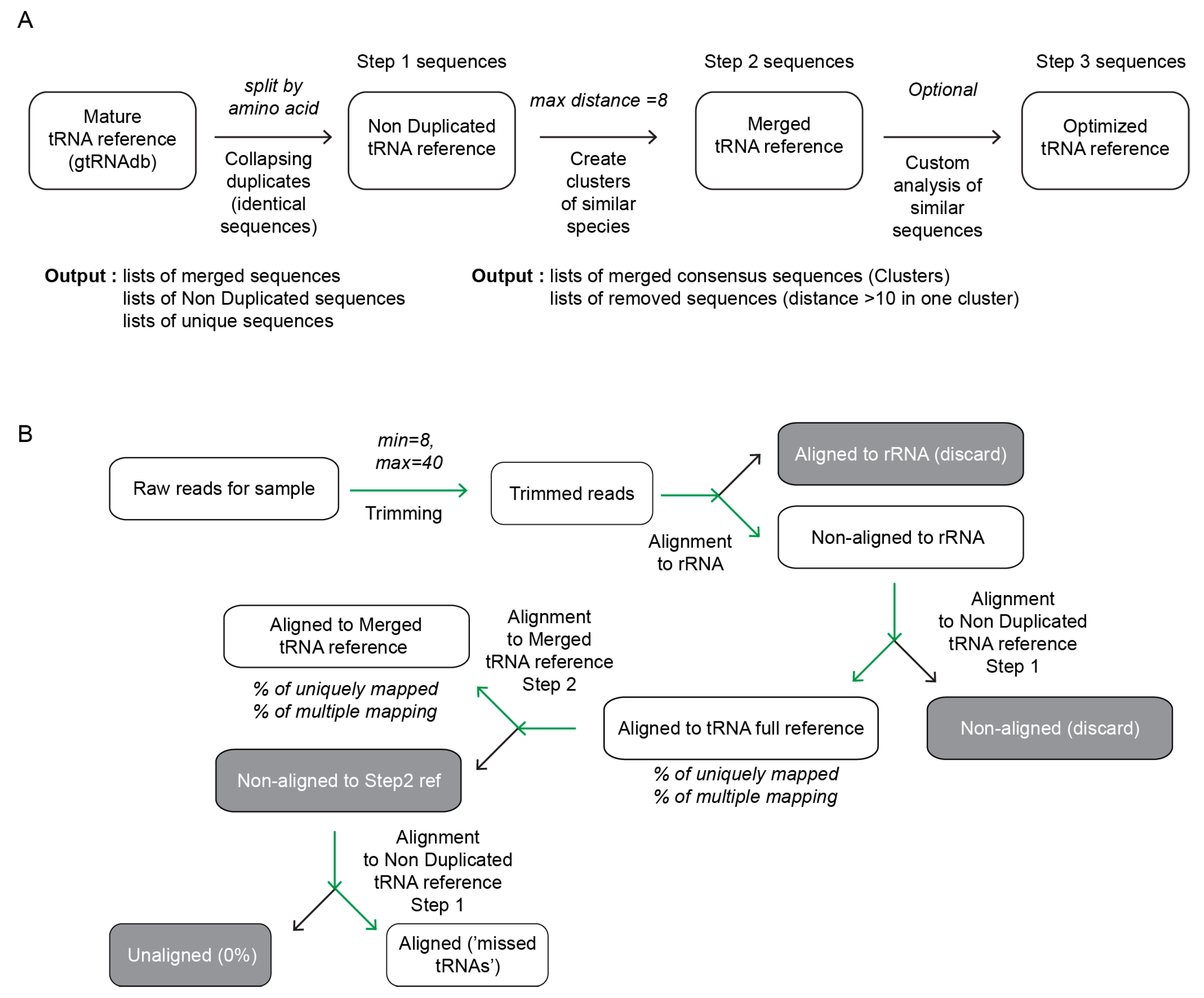
2.2.1. tRNA Reference Sequence
2.2.2. Alignment of the Experimental Datasets to Reference tRNA Sequences
2.3. Practical Guidelines for Optimization of the Reference tRNA Sequences
- Collapse full genomic tRNA dataset in collection of non-redundant sequences (automatic mode, Step 1).
- Verify the number of non-duplicated sequences (Step 1), numbers of <60 indicate almost non-redundant dataset, higher values are indication of ambiguous redundant sequences.
- Use the distance of 8 nt for genomic datasets of <250 tRNA genes (<60 Step 1 sequences) and the distance of 10 nt for larger genomic references. There may be intermediate cases for organisms having between 250 and 300 tRNA genes.
- Verify the number of optimized (Step2) sequences, values close to 40 (or less) are indication of a good quality non-redundant reference, while numbers > ~50 mean still complex and potentially redundant tRNA collection.
- Validate the optimized (Step 2) reference with experimentally obtained tRNA dataset. If proportion of uniquely mapped reads is still <90%, repeat collapsing in Step 2 with increased distance threshold.
3. Results
3.1. A Two-Steps Algorithm for tRNA Analysis
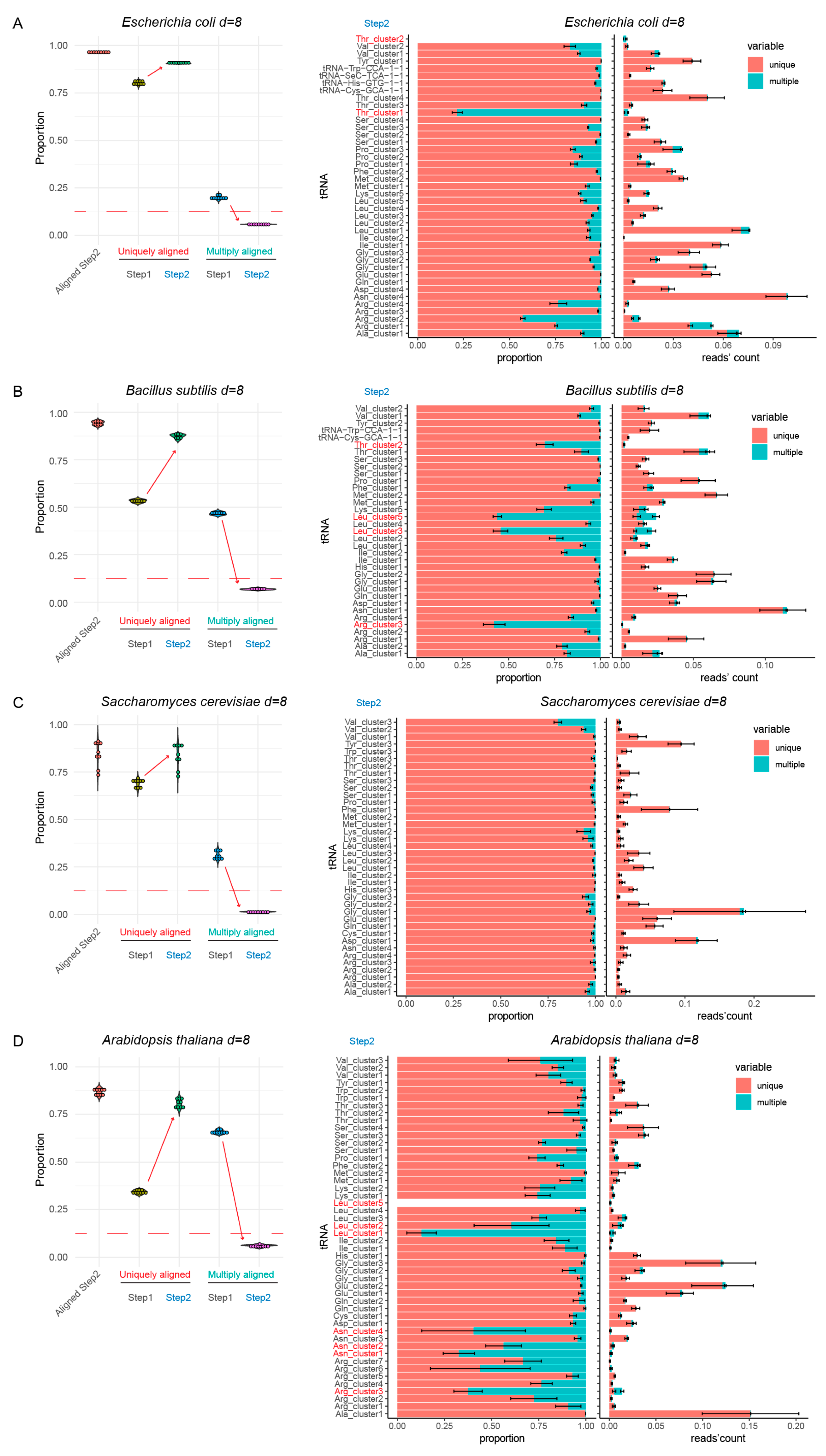
3.2. Analysis of Simple tRNA References (<100 tRNA Genes)
3.3. tRNA References of Intermediate Complexity (<300 Genes)
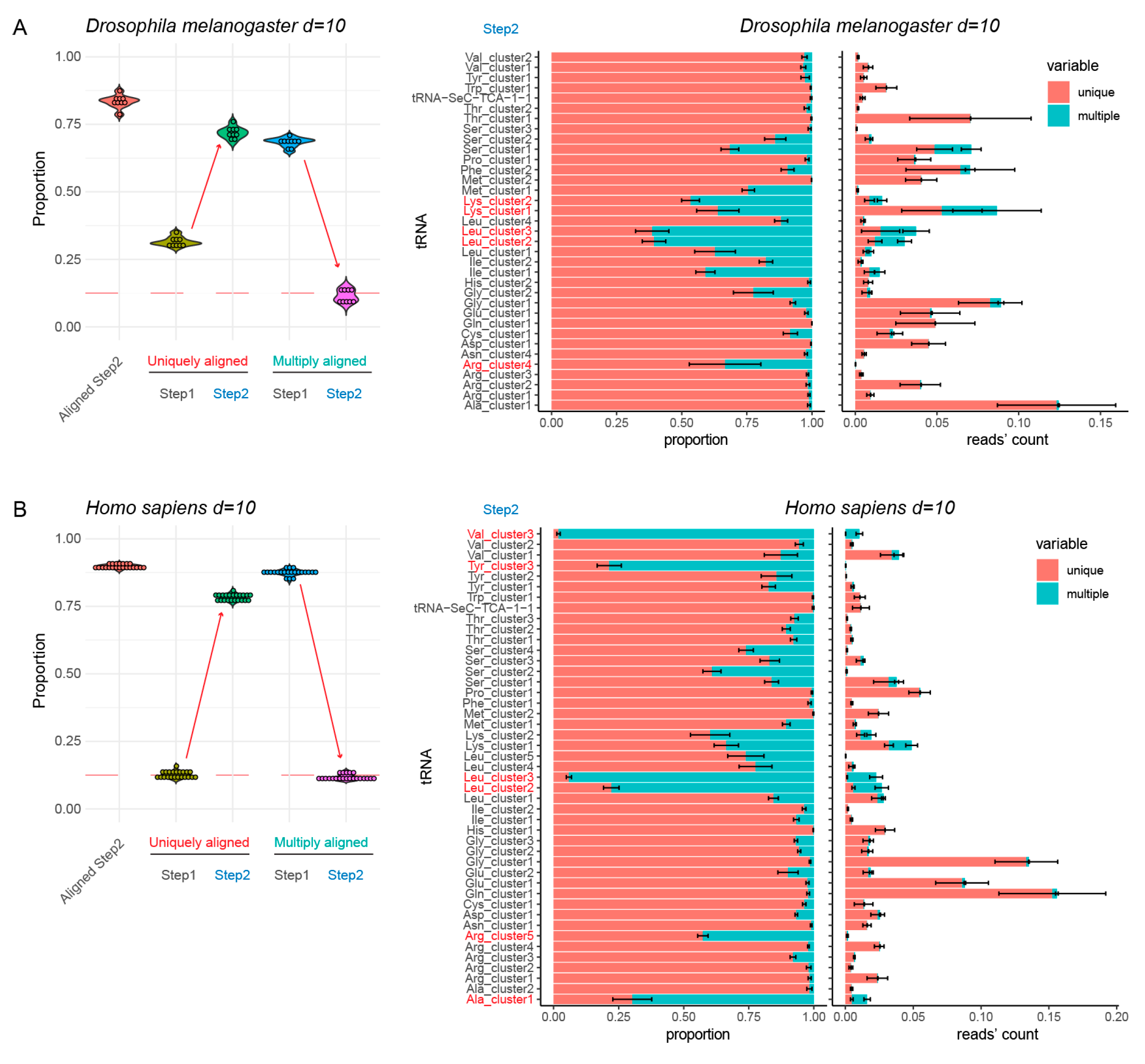
3.4. Highly Complex tRNA References (>400 Genes)
3.5. Validation of the Optimized tRNA Reference Sequences
4. Discussion
4.1. Merging of Similar tRNA Genes in a Single Reference Sequence
4.2. Representativeness of Optimized tRNA Datasets
4.3. Known Limitations and Troubleshooting
5. Conclusions
Applications in Analysis of tRNA Expression and Modifications
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berg, M.D.; Brandl, C.J. Transfer RNAs: Diversity in form and function. RNA Biol. 2020, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Burton, Z.F. Evolution of Life on Earth: tRNA, Aminoacyl-tRNA Synthetases and the Genetic Code. Life 2020, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Phizicky, E.M.; Hopper, A.K. tRNA biology charges to the front. Genes Dev. 2010, 24, 1832–1860. [Google Scholar] [CrossRef] [Green Version]
- Hori, H. Methylated nucleosides in tRNA and tRNA methyltransferases. Front Genet 2014, 5, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilusz, J.E. Controlling translation via modulation of tRNA levels. Wiley Interdiscip. Rev. RNA 2015, 6, 453–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinkard, O.; McFarland, S.; Sweet, T.; Coller, J. Quantitative tRNA-sequencing uncovers metazoan tissue-specific tRNA regulation. Nat. Commun. 2020, 11, 4104. [Google Scholar] [CrossRef]
- Clark, W.C.; Evans, M.E.; Dominissini, D.; Zheng, G.; Pan, T. tRNA base methylation identification and quantification via high-throughput sequencing. RNA 2016, 22, 1771–1784. [Google Scholar] [CrossRef] [Green Version]
- Cozen, A.E.; Quartley, E.; Holmes, A.D.; Hrabeta-Robinson, E.; Phizicky, E.M.; Lowe, T.M. ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat. Methods 2015, 12, 879–884. [Google Scholar] [CrossRef] [Green Version]
- Warren, J.M.; Salinas-Giegé, T.; Hummel, G.; Coots, N.L.; Svendsen, J.M.; Brown, K.C.; Drouard, L.; Sloan, D.B. Combining tRNA sequencing methods to characterize plant tRNA expression and post-transcriptional modification. RNA Biol. 2020, 1–15. [Google Scholar] [CrossRef]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Processing of rRNA and tRNA. In Molecular Cell Biology, 4th ed.; W. H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Drino, A.; Oberbauer, V.; Troger, C.; Janisiw, E.; Anrather, D.; Hartl, M.; Kaiser, S.; Kellner, S.; Schaefer, M.R. Production and purification of endogenously modified tRNA-derived small RNAs. RNA Biol. 2020, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kanwal, F.; Lu, C. A review on native and denaturing purification methods for non-coding RNA (ncRNA). J Chromatogr B Analyt Technol. Biomed. Life Sci. 2019, 1120, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Jacob, D.; Thüring, K.; Galliot, A.; Marchand, V.; Galvanin, A.; Ciftci, A.; Scharmann, K.; Stock, M.; Roignant, J.-Y.; Leidel, S.A.; et al. Absolute quantification of noncoding RNA by microscale thermophoresis. Angew. Chem. Int. Ed. Engl. 2019. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, D.J.; Babak, T.; Nihranz, C.; Hughes, T.R.; Engelke, D.R. Prediction and verification of mouse tRNA gene families. RNA Biol. 2009, 6, 195–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigematsu, M.; Honda, S.; Loher, P.; Telonis, A.G.; Rigoutsos, I.; Kirino, Y. YAMAT-seq: An efficient method for high-throughput sequencing of mature transfer RNAs. Nucleic Acids Res. 2017, 45, e70. [Google Scholar] [CrossRef] [PubMed]
- Erber, L.; Hoffmann, A.; Fallmann, J.; Betat, H.; Stadler, P.F.; Mörl, M. LOTTE-seq (Long hairpin oligonucleotide based tRNA high-throughput sequencing): Specific selection of tRNAs with 3’-CCA end for high-throughput sequencing. RNA Biol. 2020, 17, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Hauenschild, R.; Tserovski, L.; Schmid, K.; Thüring, K.; Winz, M.-L.; Sharma, S.; Entian, K.-D.; Wacheul, L.; Lafontaine, D.L.J.; Anderson, J.; et al. The reverse transcription signature of N-1-methyladenosine in RNA-Seq is sequence dependent. Nucleic Acids Res. 2015, 43, 9950–9964. [Google Scholar] [CrossRef] [Green Version]
- Tserovski, L.; Marchand, V.; Hauenschild, R.; Blanloeil-Oillo, F.; Helm, M.; Motorin, Y. High-throughput sequencing for 1-methyladenosine (m(1)A) mapping in RNA. Methods 2016, 107, 110–121. [Google Scholar] [CrossRef]
- Zheng, G.; Qin, Y.; Clark, W.C.; Dai, Q.; Yi, C.; He, C.; Lambowitz, A.M.; Pan, T. Efficient and quantitative high-throughput tRNA sequencing. Nat. Methods 2015, 12, 835–837. [Google Scholar] [CrossRef]
- Marchand, V.; Blanloeil-Oillo, F.; Helm, M.; Motorin, Y. Illumina-based RiboMethSeq approach for mapping of 2’-O-Me residues in RNA. Nucleic Acids Res. 2016, 44, e135. [Google Scholar] [CrossRef] [Green Version]
- Marchand, V.; Pichot, F.; Thüring, K.; Ayadi, L.; Freund, I.; Dalpke, A.; Helm, M.; Motorin, Y. Next-Generation Sequencing-Based RiboMethSeq Protocol for Analysis of tRNA 2’-O-Methylation. Biomolecules 2017, 7, 13. [Google Scholar] [CrossRef] [Green Version]
- Gogakos, T.; Brown, M.; Garzia, A.; Meyer, C.; Hafner, M.; Tuschl, T. Characterizing Expression and Processing of Precursor and Mature Human tRNAs by Hydro-tRNAseq and PAR-CLIP. Cell Rep. 2017, 20, 1463–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, P.P.; Lowe, T.M. GtRNAdb 2.0: An expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016, 44, D184–D189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, T.; Ikemura, T.; Sugahara, J.; Kanai, A.; Ohara, Y.; Uehara, H.; Kinouchi, M.; Kanaya, S.; Yamada, Y.; Muto, A.; et al. tRNADB-CE 2011: tRNA gene database curated manually by experts. Nucleic Acids Res. 2011, 39, D210–D213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Zou, Q.; Guo, J.; Ju, Y.; Wu, M.; Zeng, X.; Hong, Z. Improving tRNAscan-SE Annotation Results via Ensemble Classifiers. Mol. Inform. 2015, 34, 761–770. [Google Scholar] [CrossRef]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piatkowski, P.; Baginski, B.; Wirecki, T.K.; de Crécy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef]
- Jühling, F.; Mörl, M.; Hartmann, R.K.; Sprinzl, M.; Stadler, P.F.; Pütz, J. tRNAdb 2009: Compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009, 37, D159–D162. [Google Scholar] [CrossRef] [Green Version]
- Sajek, M.P.; Woźniak, T.; Sprinzl, M.; Jaruzelska, J.; Barciszewski, J. T-psi-C: User friendly database of tRNA sequences and structures. Nucleic Acids Res. 2020, 48, D256–D260. [Google Scholar] [CrossRef]
- Hoffmann, A.; Fallmann, J.; Vilardo, E.; Mörl, M.; Stadler, P.F.; Amman, F. Accurate mapping of tRNA reads. Bioinformatics 2018, 34, 1116–1124. [Google Scholar] [CrossRef]
- Torres, A.G.; Reina, O.; Stephan-Otto Attolini, C.; Ribas de Pouplana, L. Differential expression of human tRNA genes drives the abundance of tRNA-derived fragments. Proc. Natl. Acad. Sci. USA 2019, 116, 8451–8456. [Google Scholar] [CrossRef] [Green Version]
- Galvanin, A.; Ayadi, L.; Helm, M.; Motorin, Y.; Marchand, V. Mapping and Quantification of tRNA 2’-O-Methylation by RiboMethSeq. Methods Mol. Biol. 2019, 1870, 273–295. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujishima, K.; Kanai, A. tRNA gene diversity in the three domains of life. Front. Genet. 2014, 5, 142. [Google Scholar] [CrossRef] [Green Version]
- Wald, N.; Margalit, H. Auxiliary tRNAs: Large-scale analysis of tRNA genes reveals patterns of tRNA repertoire dynamics. Nucleic Acids Res. 2014, 42, 6552–6566. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Qian, Q.; Zhang, S.; Isaksson, L.A.; Björk, G.R. A cytosolic tRNA with an unmodified adenosine in the wobble position reads a codon ending with the non-complementary nucleoside cytidine. J. Mol. Biol. 2002, 317, 481–492. [Google Scholar] [CrossRef]
- Marchand, V.; Ayadi, L.; Ernst, F.G.M.; Hertler, J.; Bourguignon-Igel, V.; Galvanin, A.; Kotter, A.; Helm, M.; Lafontaine, D.L.J.; Motorin, Y. AlkAniline-Seq: Profiling of m7 G and m3 C RNA Modifications at Single Nucleotide Resolution. Angew. Chem. Int. Ed. Engl. 2018, 57, 16785–16790. [Google Scholar] [CrossRef]
- Marchand, V.; Pichot, F.; Neybecker, P.; Ayadi, L.; Bourguignon-Igel, V.; Wacheul, L.; Lafontaine, D.L.J.; Pinzano, A.; Helm, M.; Motorin, Y. HydraPsiSeq: A method for systematic and quantitative mapping of pseudouridines in RNA. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Lovato, M.A.; Chihade, J.W.; Schimmel, P. Translocation within the acceptor helix of a major tRNA identity determinant. EMBO J. 2001, 20, 4846–4853. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Organism | Domain | “High Confidence Set” gtRNAdb 1 | Step1 Non-Redundant tRNA Set | Step2 Optimized “Collapsed” tRNA Set (d = 8) | Step3 Validated “Optimized” tRNA Set |
|---|---|---|---|---|---|
| Plasmodium falciparum 3D7 | E | 45 | 45 | 40 | |
| Sulfolobus acidocaldarius N8 | A | 50 | 50 | 33 | |
| Haloferax volcanii DS2 | A | 52 | 46 | 46 | |
| Staphylococcus aureus subsp. aureus NCTC 8325 | B | 59 | 43 | 31 | |
| Leishmania major strain Friedlin (ASM272v2) | E | 82 | 49 | 44 | |
| Bacillus subtilis subsp. subtilis str. 168 | B | 86 | 50 | 35 | 33 |
| Escherichia coli str. K-12 substr. MG1655 | B | 89 | 48 | 41 | 39 |
| Candida albicans A20 | E | 129 | 50 | 41 | |
| Candida albicans WO-1 | E | 146 | 73 | 58 | |
| Schizosaccharomyces pombe 972h- | E | 171 | 60 | 44 | |
| Saccharomyces cerevisiae PW5 | E | 171 | 47 | 42 | |
| Candida glabrata CBS 138 | E | 189 | 45 | 38 | |
| Candida tropicalis 121 | E | 203 | 70 | 57 | |
| Saccharomyces cerevisiae P301 | E | 226 | 49 | 37 | |
| Saccharomyces cerevisiae S288c | E | 275 | 54 | 38 | 38 |
| Drosophila melanogaster (BDGP Rel. 6/dm6) | E | 290 | 76 | 37 35 (d = 10) | 34 |
| Homo sapiens (GRCh37/hg19) | E | 416 | 177 | 61 45 (d = 10) | 43 |
| Bombyx mori (Domestic silkworm ASM15162v1) | E | 435 | 115 | 44 | |
| Arabidopsis thaliana (TAIR10 Feb 2011) | E | 580 | 139 | 48 | 43 |
| Zea mays B73 (RefGen_v4 AGPv4) | 771 | 191 | 70 | ||
| Strongylocentrotus purpuratus (S. purpuratus) Mar. 2015 Spur_4.2) | E | 931 | 192 | 61 | |
| Xenopus_tropicalis_v9.1 | E | 3010 | 245 | 68 |
| Organism | tRNA Gene Number | % of Aligned Reads | Uniquely Aligned Reads 1 | Multiply Aligned Reads 1 |
|---|---|---|---|---|
| Escherichia_coli_str_K-12_substr_MG1655 | 39 | 95.87 ± 0.26 | 95.95 ± 0.23 | 4.05 ± 0.23 |
| Bacillus_subtilis_subsp_subtilis_str_168 | 33 | 94.89 ± 0.91 | 93.93 ± 0.38 | 6.07 ± 0.38 |
| Saccharomyces_cerevisiae_S288c | 38 | 84.93 ± 6.61 | 98.52 ± 0.27 | 1.48 ± 0.27 |
| Arabidopsis_thaliana_TAIR108feb2011 | 43 | 85.93 ± 2.34 | 94.08 ± 0.93 | 5.92 ± 0.93 |
| Drosophila_melanogaster_BDGP6_dm6 | 34 | 83.67 ± 2.36 | 86.88 ± 2.36 | 13.12 ± 2.36 |
| Homo_sapiens_GRCh37hg19 | 43 | 90.08 ± 0.70 | 89.15 ± 0.92 | 10.85 ± 0.92 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
PICHOT, F.; MARCHAND, V.; HELM, M.; MOTORIN, Y. Non-Redundant tRNA Reference Sequences for Deep Sequencing Analysis of tRNA Abundance and Epitranscriptomic RNA Modifications. Genes 2021, 12, 81. https://doi.org/10.3390/genes12010081
PICHOT F, MARCHAND V, HELM M, MOTORIN Y. Non-Redundant tRNA Reference Sequences for Deep Sequencing Analysis of tRNA Abundance and Epitranscriptomic RNA Modifications. Genes. 2021; 12(1):81. https://doi.org/10.3390/genes12010081
Chicago/Turabian StylePICHOT, Florian, Virginie MARCHAND, Mark HELM, and Yuri MOTORIN. 2021. "Non-Redundant tRNA Reference Sequences for Deep Sequencing Analysis of tRNA Abundance and Epitranscriptomic RNA Modifications" Genes 12, no. 1: 81. https://doi.org/10.3390/genes12010081
APA StylePICHOT, F., MARCHAND, V., HELM, M., & MOTORIN, Y. (2021). Non-Redundant tRNA Reference Sequences for Deep Sequencing Analysis of tRNA Abundance and Epitranscriptomic RNA Modifications. Genes, 12(1), 81. https://doi.org/10.3390/genes12010081