Analysis of Transcriptional Changes in Different Brassica napus Synthetic Allopolyploids

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Extraction and Library Preparation
2.3. Read Filtering, Mapping, and Analysis of Differentially Expressed Genes
2.4. Annotation of Orthologous Genomic Regions
2.5. Analyses of Expression Level Dominance and Homoeolog Expression Bias
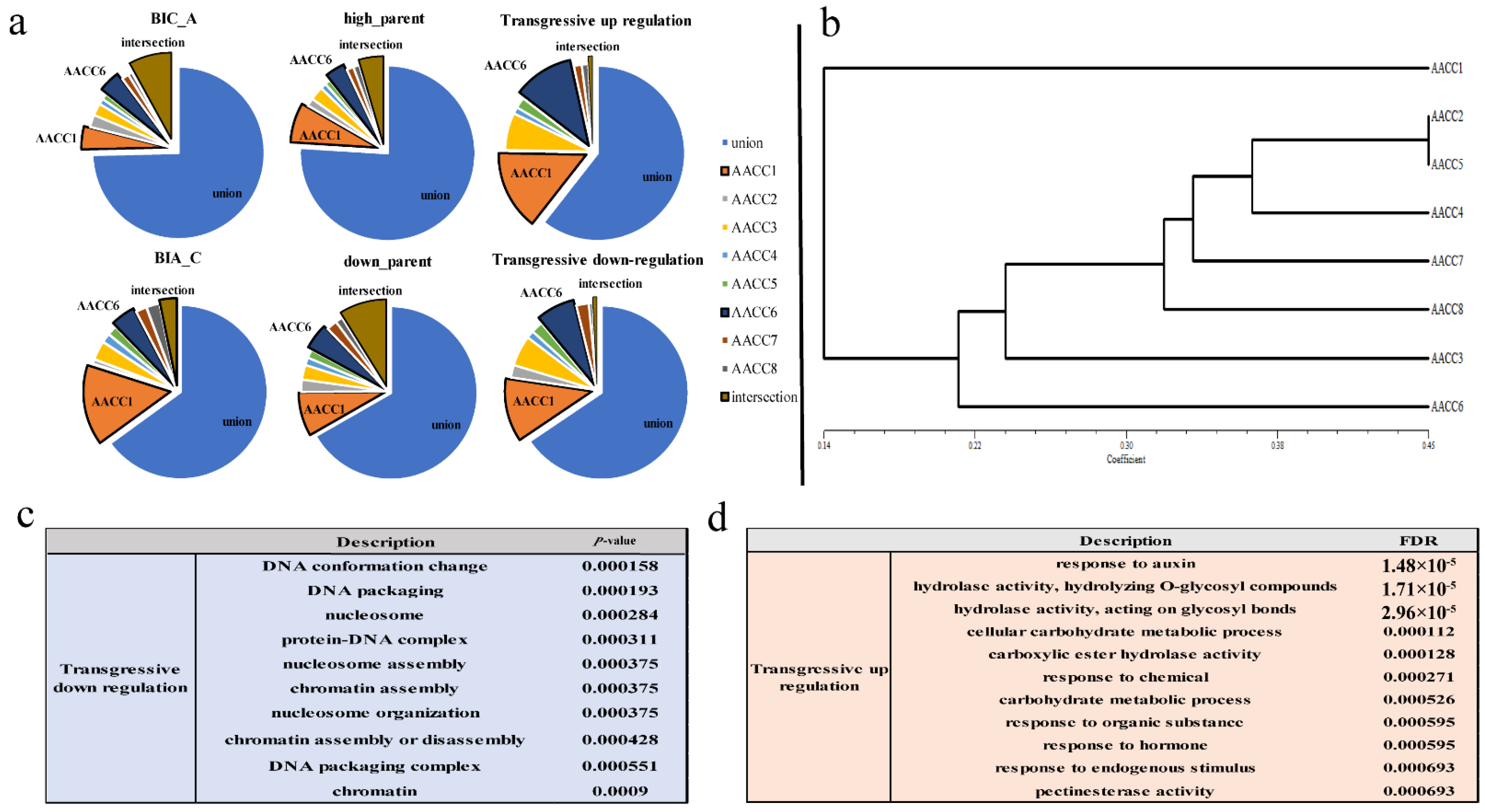
2.6. Gene Ontology (GO) Enrichment Analysis
3. Results
3.1. Transcriptome Sequencing and Read Mapping
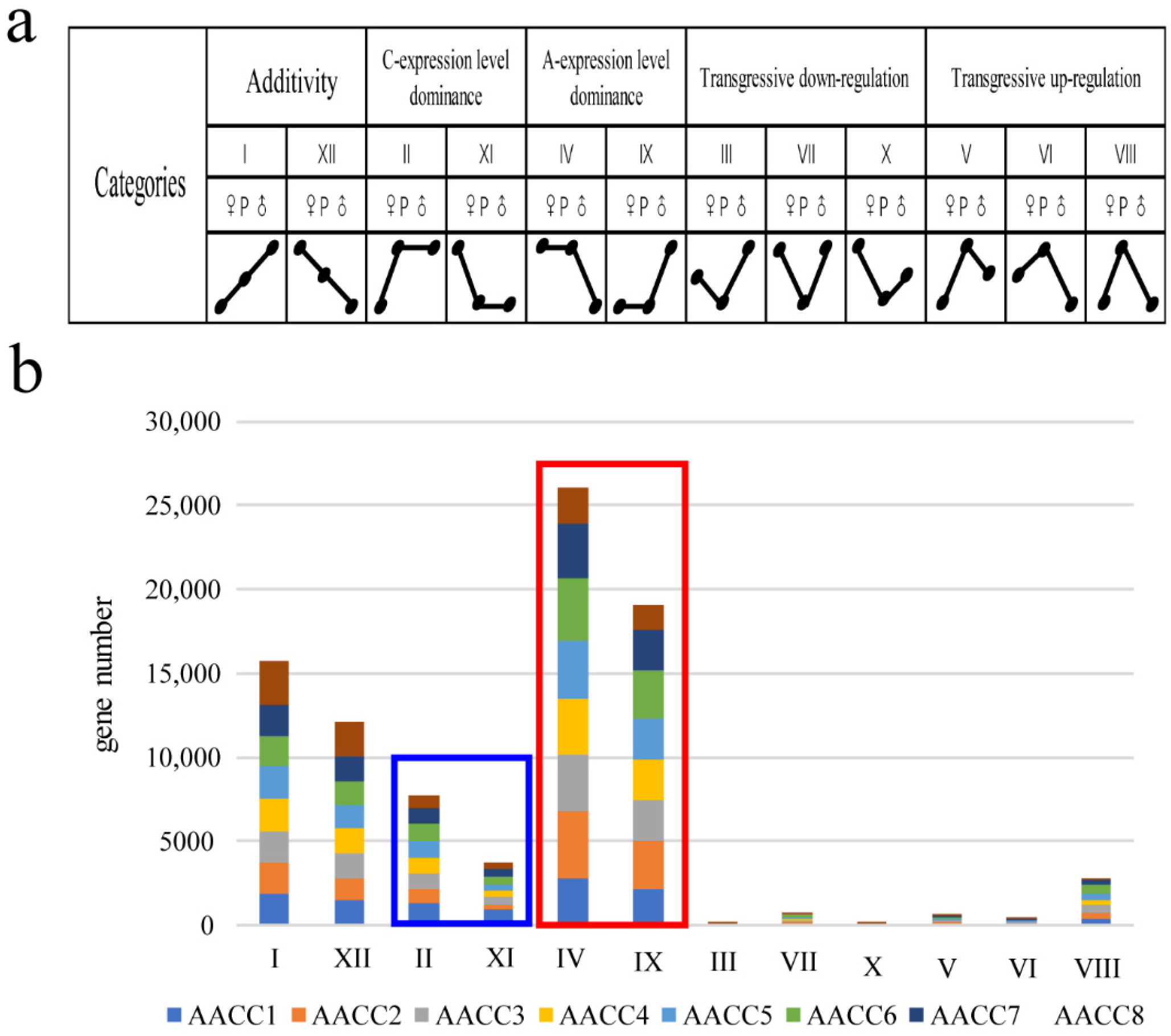
3.2. Differential, Non-Additive Gene Expressions in Synthetic Allopolyploids
3.3. Expression Level Dominance in Different Synthetic Allopolyploids
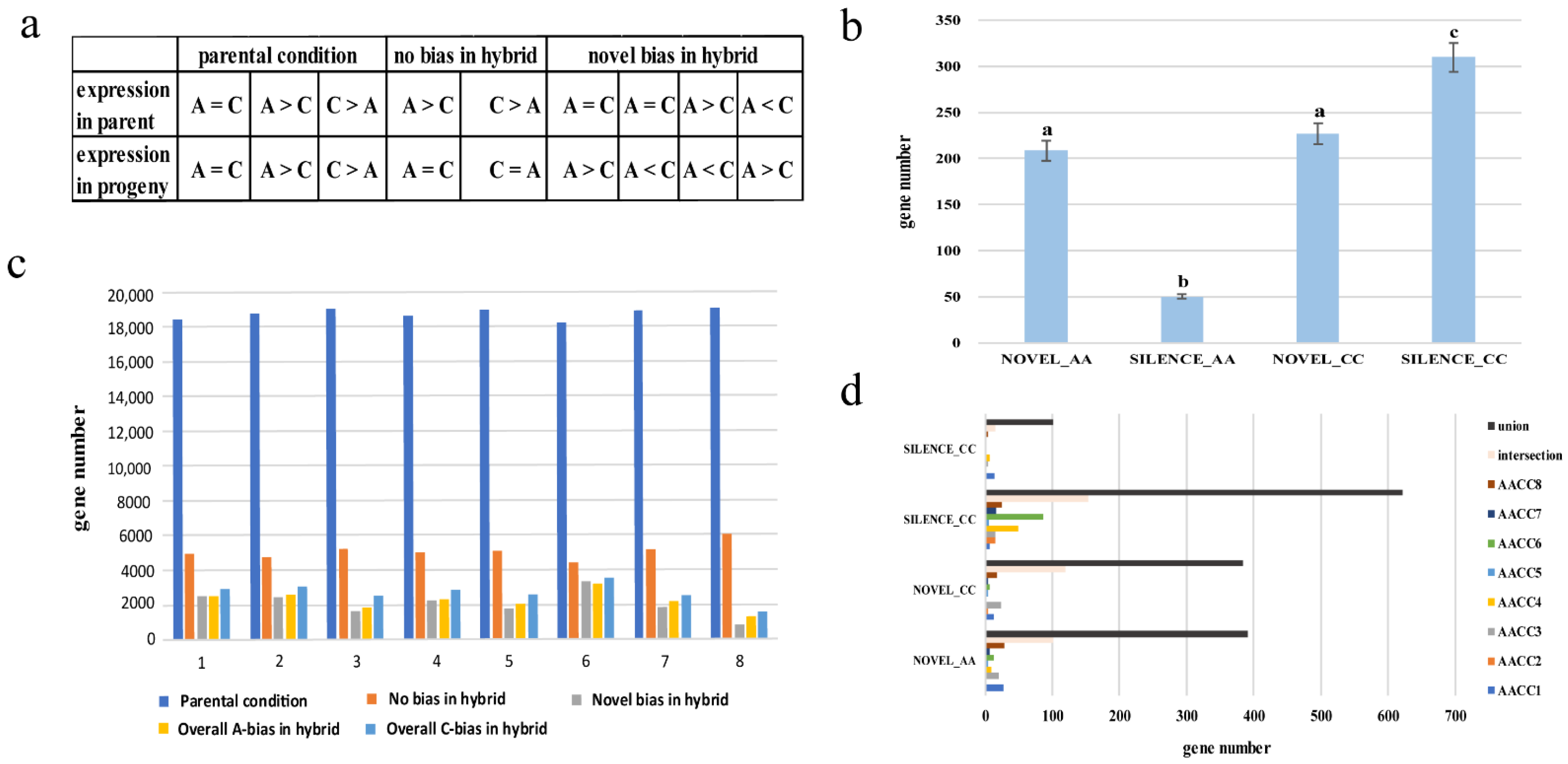
3.4. Homoeolog Expression Bias in Synthetic Allopolyploids
3.5. Novel Expression/Silencing Analysis in Synthetic Allopolyploids
3.6. Analysis of the Genes Related to Wax Synthesis and Flowering Pathway in the Eight Allopolyploids
4. Discussion
4.1. Non-Additive Expression Patterns Performance Difference in Eight Synthetic Allopolyploids
4.2. Expression Level Dominance Biased to A Genome in Eight Synthetic Allopolyploids
4.3. Homoeolog Expression Bias Performance no Difference in Eight Synthetic Allopolyploids
4.4. Differential Gene and Novel/Silenced Expression Analysis in Eight Synthetic Allopolyploids
4.5. Wax Synthesis and Flowering Pathway Among the Eight Synthetic Allotetraploids
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wendel, J.F. In Plant Molecular Evolution; Springer: Dordrecht, The Netherlands, 2000; pp. 225–249. [Google Scholar]
- Cronn, R.; Wendel, J.F. Cryptic trysts, genomic mergers, and plant speciation. New Phytol. 2004, 161, 133–142. [Google Scholar] [CrossRef] [Green Version]
- Soltis, D.E.; Soltis, P.S.; Tate, J.A. Advances in the study of polyploidy since Plant speciation. New Phytol. 2004, 161, 173–191. [Google Scholar] [CrossRef]
- Hijmans, R.J.; Gavrilenko, T.; Stephenson, S.; Bamberg, J.; Salas, A.; Spooner, D.M. Geographical and environmental range expansion through polyploidy in wild potatoes (Solanum section Petota). Glob. Ecol. Biogeogr. 2007, 16, 485–495. [Google Scholar] [CrossRef]
- Leitch, A.R.; Leitch, I.J. Genomic Plasticity and the Diversity of Polyploid Plants. Science 2008, 320, 481–483. [Google Scholar] [CrossRef]
- Nuismer, S.L.; Thompson, J.N. Plant polyploidy and non-uniform effects on insect herbivores. Proc. R. Soc. B Biol. Sci. 2001, 268, 1937–1940. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Lu, P.; Tang, K.; Osborn, T.C. Rapid genome change in synthetic polyploids of Brassica and its implications for polyploid evolution. Proc. Natl. Acad. Sci. USA 1995, 92, 7719–7723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Z.; Gaeta, R.T.; Pires, J.C. Homoeologous shuffling and chromosome compensation maintain genome balance in resynthesized allopolyploid Brassica napus. Proc. Natl. Acad. Sci. USA 2011, 108, 7908–7913. [Google Scholar] [CrossRef] [Green Version]
- Adams, K.L.; Cronn, R.; Percifield, R.; Wendel, J.F. Genes duplicated by polyploidy show unequal contributions to the transcriptome and organ-specific reciprocal silencing. Proc. Natl. Acad. Sci. USA 2003, 100, 4649–4654. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.; Ge, X.; Zhou, Y.; Li, M.; Li, Z. Cytoplasmic and genomic effects on non-meiosis-driven genetic changes in Brassica hybrids and allotetraploids from pairwise crosses of three cultivated diploids. PLoS ONE 2013, 8, e65078. [Google Scholar] [CrossRef]
- Ge, X.-H.; Ding, L.; Li, Z.-Y. Nucleolar dominance and different genome behaviors in hybrids and allopolyploids. Plant Cell Rep. 2013, 32, 1661–1673. [Google Scholar] [CrossRef]
- Wang, J.; Tian, L.; Lee, H.-S.; Wei, N.E.; Jiang, H.; Watson, B.; Madlung, A.; Osborn, T.C.; Doerge, R.W.; Comai, L.; et al. Genomewide Nonadditive Gene Regulation in Arabidopsis Allotetraploids. Genetics 2005, 172, 507–517. [Google Scholar] [CrossRef] [Green Version]
- Chelaifa, H.; Monnier, A.; Ainouche, M. Transcriptomic changes following recent natural hybridization and allopolyploidy in the salt marsh species Spartina × townsendii and Spartina anglica (Poaceae). New Phytol. 2010, 186, 161–174. [Google Scholar] [CrossRef]
- Yoo, M.J.; Szadkowski, E.; Wendel, J.F. Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity 2013, 110, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Gaeta, R.T.; Pires, J.C.; Iniguez-Luy, F.; Leon, E.; Osborn, T.C. Genomic changes in resynthesized Brassica napus and their effect on gene expression and phenotype. Plant Cell 2007, 19, 3403–3417. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Sun, S.; Hua, S.; Shen, E.; Ye, C.; Cai, D.; Timko, M.P.; Zhu, Q.; Fan, L. Analysis of transcriptional and epigenetic changes in hybrid vigor of allopolyploid Brassica napus uncovers key roles for small RNAs. Plant J. 2017, 91, 874–893. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.J. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu. Rev. Plant Biol. 2007, 58, 377–406. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, H.; Wang, J.; Sun, R.; Wu, J.; Liu, S.; Bai, Y.; Mun, J.-H.; Bancroft, I.; Cheng, F.; et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011, 43, 1035–1039. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Liu, Y.; Yang, X.; Tong, C.; Edwards, D.; Parkin, I.A.P.; Zhao, M.; Ma, J.; Yu, J.; Huang, S.; et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 2014, 5, 3930. [Google Scholar]
- An, H.; Qi, X.; Gaynor, M.L.; Hao, Y.; Gebken, S.C.; Mabry, M.E.; McAlvay, A.C.; Teakle, G.R.; Conant, G.C.; Barker, M.S.; et al. Transcriptome and organellar sequencing highlights the complex origin and diversification of allotetraploid Brassica napu. Nat. Commun. 2019, 10, 2878. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.L.; Dilkes, B.P.; McMahon, M.; Comai, L.; Nuzhdin, S.V. Homoeolog-specific retention and use in allotetraploid Arabidopsis suecica depends on parent of origin and network partners. Genome Biol. 2010, 11, 125. [Google Scholar]
- Li, A.; Liu, D.; Wu, J.; Zhao, X.; Hao, M.; Geng, S.; Yan, J.; Jiang, X.; Zhang, L.; Wu, J.; et al. mRNA and Small RNA Transcriptomes Reveal Insights into Dynamic Homoeolog Regulation of Allopolyploid Heterosis in Nascent Hexaploid Wheat. Plant Cell 2014, 26, 1878–1900. [Google Scholar] [CrossRef] [Green Version]
- Doyle, J.J.; Flagel, L.E.; Paterson, A.H.; Rapp, R.A.; Soltis, D.E.; Soltis, P.S.; Wendel, J.F. Evolutionary Genetics of Genome Merger and Doubling in Plants. Annu. Rev. Genet. 2008, 42, 443–461. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Kang, L.; Li, P.; Li, Z. Review on new germplasm development in Brassica napus through wide hybridizations in China. Chin. J. Oil Crop Sci. 2016, 38, 691. [Google Scholar]
- Ren, J.P.; Dickson, M.H.; Earle, E.D. Improved resistance to bacterial soft rot by protoplast fusion between Brassica rapa and B. oleracea. Theor. Appl. Genet. 2000, 100, 810–819. [Google Scholar]
- Yoo, M.J.; Liu, X.; Pires, J.C.; Soltis, P.S.; Soltis, D.E. Nonadditive gene expression in polyploids. Annu. Rev. Genet. 2014, 48, 485–517. [Google Scholar] [CrossRef]
- Zhang, D.; Pan, Q.; Tan, C.; Zhu, B.; Ge, X.; Shao, Y.; Li, Z. Genome-Wide Gene Expressions Respond Differently to A-subgenome Origins in Brassica napus Synthetic Hybrids and Natural Allotetraploid. Front. Plant Sci. 2016, 7, 1508. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Zou, J.; Meng, J.; Mei, S.; Wang, J. Tracing the transcriptomic changes in synthetic Trigenomic allohexaploids of Brassica using an RNA-Seq approach. PLoS ONE 2013, 8, e68883. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Lin, L.; Xu, M.; Chen, P.; Liu, D.; Sun, Q.; Ran, L.; Wang, Y. Homoeolog expression bias and expression level dominance in resynthesized allopolyploid Brassica napus. BMC Genom. 2018, 19, 586. [Google Scholar]
- Wei, Y.; Li, F.; Zhang, S.; Zhang, S.; Zhang, H.; Sun, R. Characterization of Interspecific Hybrids between Flowering Chinese Cabbage and Chinese Kale. Agronomy 2018, 8, 258. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Kalyna, M.; Simpson, C.G.; Syed, N.H.; Lewandowska, D.; Marquez, Y.; Kusenda, B.; Marshall, J.; Fuller, J.; Cardle, L.; McNicol, J.; et al. Alternative splicing and nonsense-mediated decay modulate expression of important regulatory genes in Arabidopsis. Nucleic Acids Res. 2012, 40, 2454–2469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Ng, D.W.-K.; Zhang, C.; Comai, L.; Ye, W.; Chen, Z.J. Cis- and trans-regulatory divergence between progenitor species determines gene-expression novelty in Arabidopsis allopolyploids. Nat. Commun. 2012, 3, 950. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Pan, Q.; Cui, C.; Tan, C.; Ge, X.; Shao, Y.; Li, Z. Genome-specific differential gene expressions in resynthesized Brassica allotetraploids from pair-wise crosses of three cultivated diploids revealed by RNA-seq. Front. Plant Sci. 2015, 6, 957. [Google Scholar]
- Grover, C.E.; Gallagher, J.P.; Szadkowski, E.; Yoo, M.J.; Flagel, L.E.; Wendel, J.F. Homoeolog expression bias and expression level dominance in allopolyploids. New Phytol. 2012, 196, 966–971. [Google Scholar] [CrossRef]
- Nicolas, S.D.; Leflon, M.; Liu, Z.; Eber, F.; Chelysheva, L.; Coriton, O.; Chèvre, A.M.; Jenczewski, E. Chromosome ‘speed dating’ during meiosis of polyploid Brassica hybrids and haploids. Cytogenet. Genome Res. 2008, 120, 331–338. [Google Scholar] [CrossRef]
- Aarts, M.G.; Keijzer, C.J.; Stiekema, W.J.; Pereira, A. Molecular characterization of the CER1 gene of arabidopsis involved in epicuticular wax biosynthesis and pollen fertility. Plant Cell 1995, 7, 2115. [Google Scholar]
- Rowland, O.; Lee, R.; Franke, R.; Schreiber, L.; Kunst, L. The CER3 wax biosynthetic gene from Arabidopsis thaliana is allelic to WAX2/YRE/FLP1. FEBS Lett. 2007, 581, 3538–3544. [Google Scholar]
- Chen, X. Cloning and Characterization of the WAX2 Gene of Arabidopsis Involved in Cuticle Membrane and Wax Production. Plant Cell Online 2003, 15, 1170–1185. [Google Scholar] [CrossRef] [Green Version]
- Bernard, A.; Domergue, F.; Pascal, S.; Jetter, R.; Renne, C.; Faure, J.-D.; Haslam, R.P.; Napier, J.A.; Lessire, R.; Joubès, J. Reconstitution of Plant Alkane Biosynthesis in Yeast Demonstrates That Arabidopsis ECERIFERUM1 and ECERIFERUM3 Are Core Components of a Very-Long-Chain Alkane Synthesis Complex. Plant Cell 2012, 24, 3106–3118. [Google Scholar]
- Chagué, V.; Just, J.; Mestiri, I.; Balzergue, S.; Tanguy, A.-M.; Huneau, C.; Huteau, V.; Belcram, H.; Coriton, O.; Jahier, J.; et al. Genome-wide gene expression changes in genetically stable synthetic and natural wheat allohexaploids. New Phytol. 2010, 187, 1181–1194. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, Y.; Zhu, B.; Fang, T.; Fang, Y.; Wang, Y. Digital gene expression analysis of gene expression differences within Brassica diploids and allopolyploids. BMC Plant Biol. 2015, 15, 22. [Google Scholar] [CrossRef] [Green Version]
- He, L.-Q.; Tang, R.-H.; Jiang, J.; Xiong, F.-Q.; Huang, Z.-P.; Wu, H.-N.; Gao, Z.-K.; Zhong, R.-C.; He, X.-H.; Han, Z.-Q. Rapid gene expression change in a novel synthesized allopolyploid population of cultivated peanut×Arachis doigoi cross by cDNA-SCoT and HFO-TAG technique. J. Integr. Agric. 2017, 16, 1093–1102. [Google Scholar] [CrossRef] [Green Version]
- Gaeta, R.T.; Yoo, S.-Y.; Pires, J.C.; Doerge, R.W.; Chen, Z.J.; Osborn, T.C. Analysis of Gene Expression in Resynthesized Brassica napus Allopolyploids Using Arabidopsis 70mer Oligo Microarrays. PLoS ONE 2009, 4, e4760. [Google Scholar] [CrossRef] [Green Version]
- Rapp, R.A.; Udall, J.A.; Wendel, J.F. Genomic expression dominance in allopolyploids. BMC Biol. 2009, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Flagel, L.E.; Wendel, J.F. Evolutionary rate variation, genomic dominance and duplicate gene expression evolution during allotetraploid cotton speciation. New Phytol. 2010, 186, 184–193. [Google Scholar] [CrossRef]
- Qi, B.; Huang, W.; Zhu, B.; Zhong, X.; Guo, J.; Zhao, N.; Xu, C.; Zhang, H.; Pang, J.; Han, F.; et al. Global transgenerational gene expression dynamics in two newly synthesized allohexaploid wheat (Triticum aestivum) lines. BMC Biol. 2012, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Hollister, J.D.; Gaut, B.S. Epigenetic silencing of transposable elements: A trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 2009, 19, 1419–1428. [Google Scholar] [CrossRef] [Green Version]
- Freeling, M.; Woodhouse, M.R.; Subramaniam, S.; Turco, G.; Lisch, D.; Schnable, J.C. Fractionation mutagenesis and similar consequences of mechanisms removing dispensable or less-expressed DNA in plants. Curr. Opin. Plant Biol. 2012, 15, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J.; Pikaard, C.S. Epigenetic silencing ofRNA polymerase I transcription: A role forDNAmethylation and histone modification in nucleolar dominance. Genes Dev. 1997, 11, 2124–2136. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.J.; Pikaard, C.S. Transcriptional analysis of nucleolar dominance in polyploid plants: Biased expression/silencing of progenitor rRNA genes is developmentally regulated in Brassica. Proc. Natl. Acad. Sci. USA 1997, 94, 3442–3447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Zhu, M.; Qiao, H.; Li, F.; Zhang, S.; Zhang, S.; Zhang, H.; Sun, R. Characterization of interspecific hybrids between flowering Chinese cabbage and broccoli. Sci. Hortic. 2018, 240, 552–557. [Google Scholar] [CrossRef]
- Nah, G.; Jeffrey Chen, Z. Tandem duplication of the FLC locus and the origin of a new gene in Arabidopsis related species and their functional implications in allopolyploids. New Phytol. 2010, 186, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Szadkowski, E.; Eber, F.; Huteau, V.; Lodé, M.; Huneau, C.; Belcram, H.; Coriton, O.; Manzanares-Dauleux, M.J.; Delourme, R.; King, G.J.; et al. The first meiosis of resynthesized Brassica napus, a genome blender. New Phytol. 2010, 186, 102–112. [Google Scholar] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Y.; Li, G.; Zhang, S.; Zhang, S.; Zhang, H.; Sun, R.; Zhang, R.; Li, F. Analysis of Transcriptional Changes in Different Brassica napus Synthetic Allopolyploids. Genes 2021, 12, 82. https://doi.org/10.3390/genes12010082
Wei Y, Li G, Zhang S, Zhang S, Zhang H, Sun R, Zhang R, Li F. Analysis of Transcriptional Changes in Different Brassica napus Synthetic Allopolyploids. Genes. 2021; 12(1):82. https://doi.org/10.3390/genes12010082
Chicago/Turabian StyleWei, Yunxiao, Guoliang Li, Shujiang Zhang, Shifan Zhang, Hui Zhang, Rifei Sun, Rui Zhang, and Fei Li. 2021. "Analysis of Transcriptional Changes in Different Brassica napus Synthetic Allopolyploids" Genes 12, no. 1: 82. https://doi.org/10.3390/genes12010082
APA StyleWei, Y., Li, G., Zhang, S., Zhang, S., Zhang, H., Sun, R., Zhang, R., & Li, F. (2021). Analysis of Transcriptional Changes in Different Brassica napus Synthetic Allopolyploids. Genes, 12(1), 82. https://doi.org/10.3390/genes12010082