
Catalogue for Transmission Genetics in Arabs (CTGA) Database: Analysing Lebanese Data on Genetic Disorders

 ,
,
Abstract
:1. Introduction
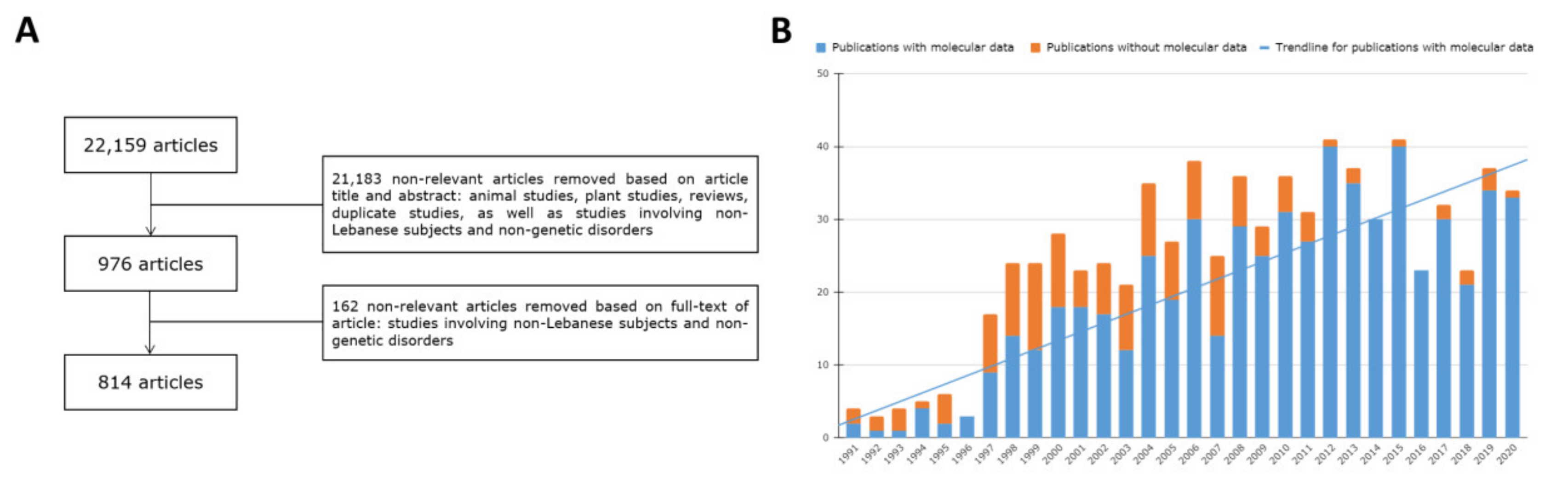
2. Materials and Methods
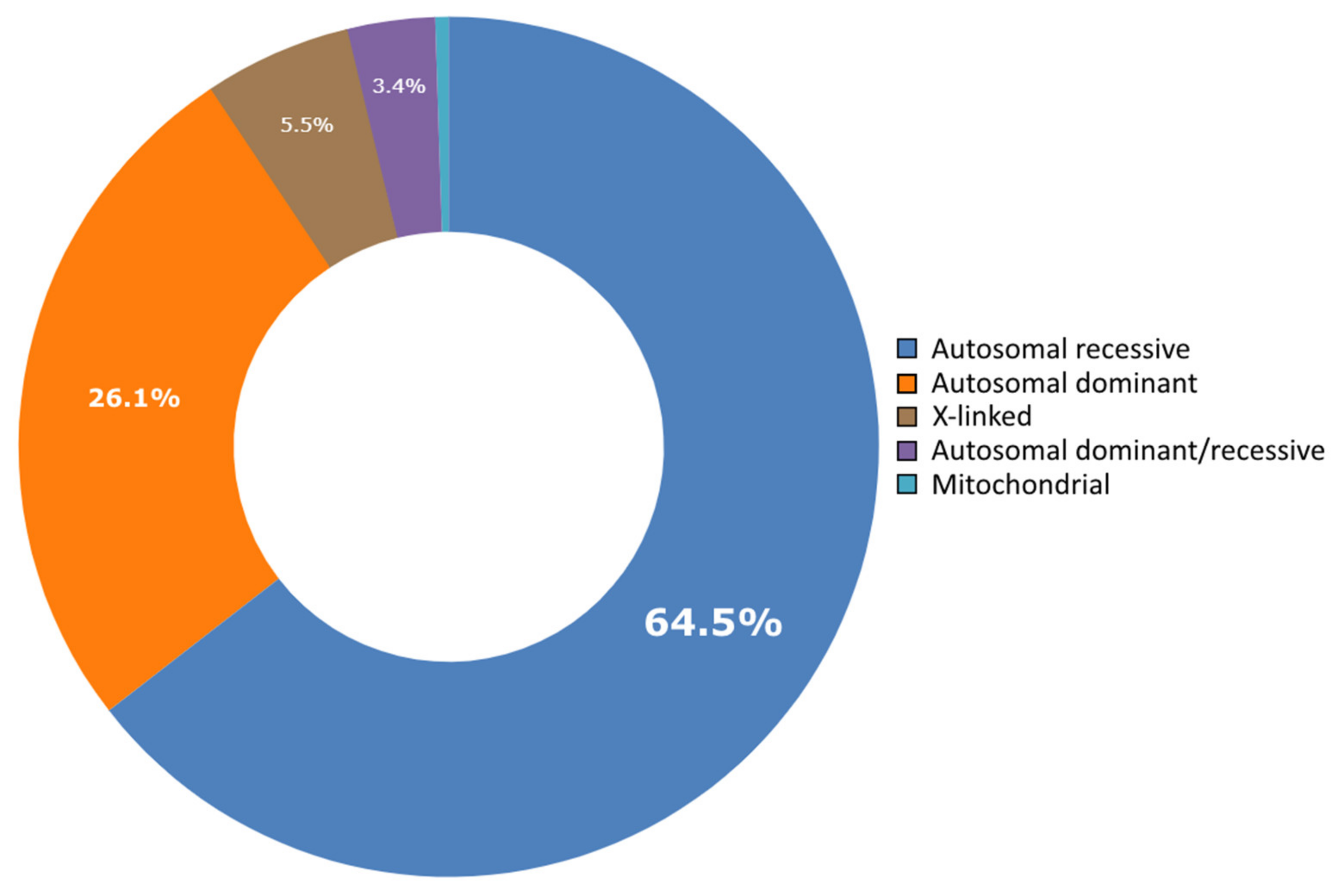
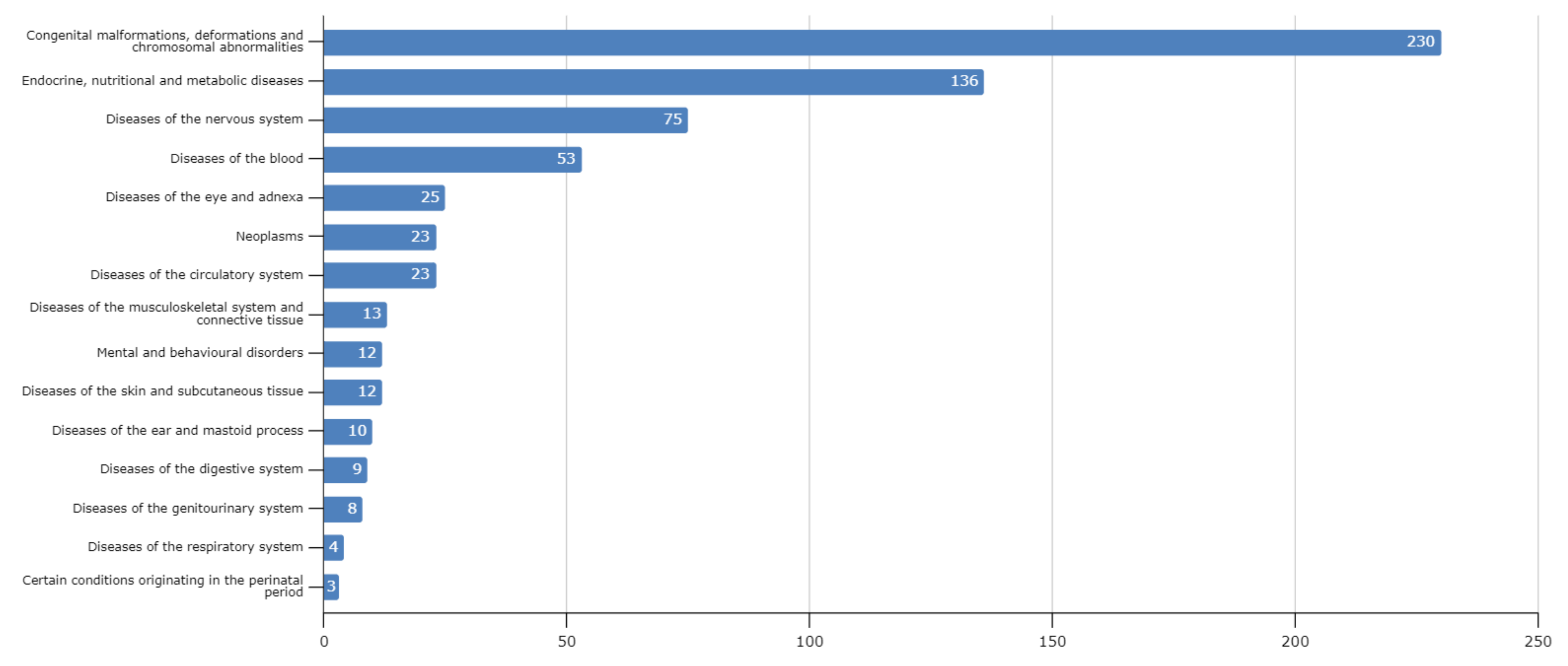
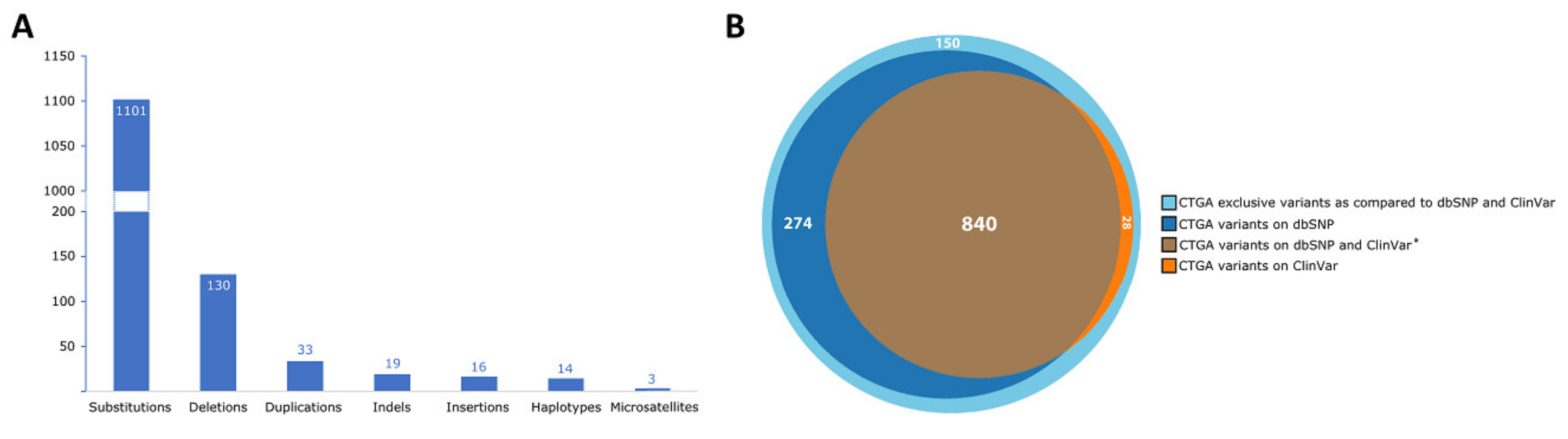
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- UNPFA. World Population Dashboard: Lebanon. Available online: https://www.unfpa.org/data/world-population/LB (accessed on 23 August 2021).
- UNHCR. 2020 Year-End Results. Available online: https://reporting.unhcr.org/lebanon (accessed on 23 August 2021).
- Tabar, P.; Denison, A. Diaspora Policies, Consular Services and Social Protection for Lebanese Citizens Abroad. In Migration and Social Protection in Europe and Beyond (Volume 3): A Focus on Non-EU Sending States; Lafleur, J.-M., Vintila, D., Eds.; Springer International Publishing: Cham, Germany, 2020; pp. 199–215. [Google Scholar]
- Christianson, A.; Howson, C.P.; Modell, B. March of Dimes: Global Report on Birth Defects; March of Dimes Birth Defects Foundation: White Plains, NY, USA, 2006. [Google Scholar]
- Al-Gazali, L.; Hamamy, H.; Al-Arrayad, S. Genetic disorders in the Arab world. BMJ 2006, 333, 831–834. [Google Scholar] [CrossRef] [Green Version]
- Der Kaloustian, V.M. Genetic Disorders in Lebanon. In Genetic Disorders among Arab Populations; Teebi, A.S., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 377–441. [Google Scholar]
- Al-Gazali, L.; Hamamy, H. Consanguinity and dysmorphology in Arabs. Hum. Hered. 2014, 77, 93–107. [Google Scholar] [CrossRef]
- Nakouzi, G.; Kreidieh, K.; Yazbek, S. A review of the diverse genetic disorders in the Lebanese population: Highlighting the urgency for community genetic services. J. Community Genet. 2015, 6, 83–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issam Khneisser, S.M.A.; Megarbane, A. Genetic Disorders in Lebanon: Challenges and Opportunities. In Genomics and Health in the Developing World; Kumar, D., Ed.; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Der Kaloustian, V.M. Genetic diseases in Lebanon. Am. J. Med. Genet. 1986, 36, 65–68. [Google Scholar] [CrossRef]
- CAGS. Catalogue for Transmission Genetics in Arabs Database. Available online: https://cags.org.ae/en/ctga-overview (accessed on 23 August 2021).
- Pipkin, A.C.; Pipkin, S.B. A pedigree of generalized lentigo. J. Hered. 1950, 41, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Bissar-Tadmouri, N.; Tadmouri, G.O. Bibliometric analyses of biomedical research outputs in Lebanon and the United Arab Emirates (1988–2007). Saudi Med. J. 2009, 30, 130–139. [Google Scholar] [PubMed]
- Megarbane, A.; Ghanem, I.; Romana, S.; Gosset, P.; Caillaud, C. Congenital contractures, short stature, abnormal face, microcephaly, scoliosis, hip dislocation, and severe psychomotor retardation in two unrelated girls. A new MCA/MR syndrome? Genet. Couns. 2002, 13, 123–131. [Google Scholar] [PubMed]
- Megarbane, A.; Haddad-Zebouni, S.; Nabbout, R.; Khoury, A.H.; Traboulsi, E.I. Microcephaly, colobomatous microphthalmia, short stature, and severe psychomotor retardation in two male cousins: A new MCA/MR syndrome? Am. J. Med. Genet. 1999, 83, 82–87. [Google Scholar] [CrossRef]
- Wakim, V.; Nair, P.; Delague, V.; Bizzari, S.; Al-Ali, M.T.; Castro, C.; Alicia, G.; Stephany, E.-H.; André, M.M. SOX11-related syndrome: Report on a new case and review. Clin. Dysmorphol. 2021, 30, 44–49. [Google Scholar] [CrossRef]
- Hana, S.; Karthik, D.; Shan, J.; El Hayek, S.; Chouchane, L.; Megarbane, A. A Report on a Family with TMTC3-Related Syndrome and Review. Case Rep. Med. 2020, 2020, 7163038. [Google Scholar] [CrossRef]
- Bizzari, S.; El-Bazzal, L.; Nair, P.; Younan, A.; Stora, S.; Mehawej, C.; El-Hayek, S.; Delague, V.; Mégarbané, A. Recessive marfanoid syndrome with herniation associated with a homozygous mutation in Fibulin-3. Eur. J. Med. Genet. 2020, 63, 103869. [Google Scholar] [CrossRef] [PubMed]
- Megarbane, A.; Rassi, S.; Estephan, F.; Kouba-Hreich, E. Post-natal short stature, short limbs, brachydactyly, facial abnormalities, and delayed bone age: A new syndrome? Am. J. Med. Genet. Part A 2004, 125A, 57–60. [Google Scholar] [CrossRef]
- Oniya, O.; Neves, K.; Ahmed, B.; Konje, J.C. A review of the reproductive consequences of consanguinity. Eur. J. Obstet. Gynecol. Reprod. Biol. 2019, 232, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.; Obaid, T.; Tadmouri, G.O. Genetic Disorders in Qatar: A CTGA Perspective. In Genetic Disorders in the Arab World: Qatar; Taleb Al Ali, M., Ed.; CAGS: Dubai, United Arab Emirates, 2012; Volume 4. [Google Scholar]
- Farah, R.A.; Nair, P.; Koueik, J.; Yammine, T.; Khalifeh, H.; Korban, R.; Agnes, C.; Claudia, K.; Catherine, D.-D.; Eliane, C.; et al. Clinical and Genetic Features of Patients with Fanconi Anemia in Lebanon and Report on Novel Mutations in the FANCA and FANCG Genes. J. Pediatr. Hematol./Oncol. 2021, 43, e727–e735. [Google Scholar] [CrossRef] [PubMed]
- Farra, C.; Badra, R.; Fares, F.; Muwakkit, S.; Dbaibo, G.; Dabbous, I.; Ashkar, H.; Mounsef, C.; Abboud, M.R. Alpha thalassemia allelic frequency in Lebanon. Pediatr. Blood Cancer 2015, 62, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Inati, A.; Zeineh, N.; Isma’eel, H.; Koussa, S.; Gharzuddine, W.; Taher, A. Beta-thalassemia: The Lebanese experience. Clin. Lab. Haematol. 2006, 28, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Jelwan, Y.A.; Asbeutah, A.A.A.; Welty, F.K. Comprehensive Review of Cardiovascular Diseases, Diabetes, and Hypercholesterolemia in Lebanon. Cardiol. Rev. 2020, 28, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Maddirevula, S.; Shamseldin, H.E.; Sirr, A.; AlAbdi, L.; Lo, R.S.; Ewida, N.; Al-Qahtani, M.; Hashem, M.; Abdulwahab, F.; Aboyousef, O.; et al. Exploiting the Autozygome to Support Previously Published Mendelian Gene-Disease Associations: An Update. Front. Genet. 2020, 11, 580484. [Google Scholar] [CrossRef]
- Romdhane, L.; Mezzi, N.; Hamdi, Y.; El-Kamah, G.; Barakat, A.; Abdelhak, S. Consanguinity and Inbreeding in Health and Disease in North African Populations. Annu. Rev. Genom. Hum. Genet. 2019, 20, 155–179. [Google Scholar] [CrossRef]
- Alkuraya, F.S. Autozygome decoded. Genet. Med. 2010, 12, 765–771. [Google Scholar] [CrossRef]
- Megarbane, A. Clinical genetics revisited: Effect of new techniques (next-generation sequencing, comparative genomic hybridization) on previous diagnoses. Middle East. J. Med. Genet. 2018, 7, 1–6. [Google Scholar]
- Lim, S.C.; Smith, K.R.; Stroud, D.; Compton, A.; Tucker, E.; Dasvarma, A.; Gandolfo, L.C.; Marum, J.E.; McKenzie, M.; Peters, H.L.; et al. A founder mutation in PET100 causes isolated complex IV deficiency in Lebanese individuals with Leigh syndrome. Am. J. Hum. Genet. 2014, 94, 209–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalkh, N.; Corbani, S.; Haidar, Z.; Hamdan, N.; Farah, E.; Abou Ghoch, J.; Ghosn, R.; Salem, N.; Fawaz, A.; Khayat, C.D.; et al. The added value of WES reanalysis in the field of genetic diagnosis: Lessons learned from 200 exomes in the Lebanese population. BMC Med. Genom. 2019, 12, 11. [Google Scholar] [CrossRef] [Green Version]
- Mansour, H.; Sabbagh, S.; Bizzari, S.; El-Hayek, S.; Chouery, E.; Gambarini, A.; Gencik, M.; Mégarbané, A. The Lebanese Allele in the PET100 Gene: Report on Two New Families with Cytochrome c Oxidase Deficiency. J. Pediatr. Genet. 2019, 8, 172–178. [Google Scholar] [CrossRef]
- Riley, L.G.; Cowley, M.J.; Gayevskiy, V.; Minoche, A.E.; Puttick, C.; Thorburn, D.; Rius, R.; Compton, A.; Menezes, M.J.; Bhattacharya, K.; et al. The diagnostic utility of genome sequencing in a pediatric cohort with suspected mitochondrial disease. Genet. Med. 2020, 22, 1254–1261. [Google Scholar] [CrossRef]
- Fahed, A.C.; Safa, R.M.; Haddad, F.F.; Bitar, F.F.; Andary, R.R.; Arabi, M.T.; Azar, S.T.; Nemer, G. Homozygous familial hypercholesterolemia in Lebanon: A genotype/phenotype correlation. Mol. Genet. Metab. 2011, 102, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Fahed, A.C.; Bitar, F.F.; Khalaf, R.I.; Moubarak, E.M.; Azar, S.T.; Nemer, G.M. The Lebanese allele at the LDLR in normocholesterolemic people merits reconsideration of genotype phenotype correlations in familial hypercholesterolemia. Endocrine 2012, 42, 445–448. [Google Scholar] [CrossRef]
- Dos Santos, J.E.; Zago, M.A. Familial hypercholesterolemia in Brazil. Atheroscler. Suppl. 2003, 4, 1–2. [Google Scholar] [CrossRef]
- Fahed, A.C.; Khalaf, R.; Salloum, R.; Andary, R.R.; Safa, R.; El-Rassy, I.; Moubarak, E.; Azar, S.T.; Bitar, F.F.; Nemer, G. Variable expressivity and co-occurrence of LDLR and LDLRAP1 mutations in familial hypercholesterolemia: Failure of the dominant and recessive dichotomy. Mol. Genet. Genom. Med. 2016, 4, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Fahed, A.C.; Shibbani, K.; Andary, R.R.; Arabi, M.T.; Habib, R.H.; Nguyen, D.D.; Haddad, F.F.; Moubarak, E.; Nemer, G.; Azar, S.T.; et al. Premature Valvular Heart Disease in Homozygous Familial Hypercholesterolemia. Cholesterol 2017, 2017, 3685265. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.O.; Gibbs, J.R.; Megarbane, A.; Urtizberea, J.A.; Hernandez, D.G.; Foley, A.R.; Arepalli, S.; Pandraud, A.; Simón-Sánchez, J.; Clayton, P.; et al. Exome sequencing reveals riboflavin transporter mutations as a cause of motor neuron disease. Brain 2012, 135, 2875–2882. [Google Scholar] [CrossRef] [PubMed]
- Srour, M.; Putorti, M.L.; Schwartzentruber, J.; Bolduc, V.; Shevell, M.I.; Poulin, C.; O’ferrall, E.; Buhas, D.; Majewski, J.; Brais, B. Mutations in riboflavin transporter present with severe sensory loss and deafness in childhood. Muscle Nerve 2014, 50, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Simha, V.; Oral, E.A.; Moran, S.A.; Gorden, P.; O’Rahilly, S.; Zaidi, Z.; Gurakan, F.; Arslanian, S.A.; Klar, A.; et al. Phenotypic and genetic heterogeneity in congenital generalized lipodystrophy. J. Clin. Endocrinol. Metab. 2003, 88, 4840–4847. [Google Scholar] [CrossRef]
- Van Maldergem, L.; Magre, J.; Khallouf, T.E.; Gedde-Dahl, T.; Delépine, M.; Trygstad, O.; Seemanova, E.; Stephenson, T.; Albott, C.S.; Bonnici, F.; et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J. Med. Genet. 2002, 39, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.Q.; Ward, L.M.; Liu, S.; Lu, Y.B.; Xie, Y.X.; Yuan, B.Z.; Yu, X.J.; Rauch, F.; Davis, S.I.; Zhang, S.B.; et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat. Genet. 2006, 38, 1310–1315. [Google Scholar] [CrossRef]
- Gannage-Yared, M.H.; Makrythanasis, P.; Chouery, E.; Sobacchi, C.; Mehawej, C.; Santoni, F.A.; Guipponi, M.; Antonarakis, S.E.; Hamamy, H.; Mégarbané, A. Exome sequencing reveals a mutation in DMP1 in a family with familial sclerosing bone dysplasia. Bone 2014, 68, 142–145. [Google Scholar] [CrossRef]
- Lorenz-Depiereux, B.; Bastepe, M.; Benet-Pages, A.; Amyere, M.; Wagenstaller, J.; Müller-Barth, U.; Badenhoop, K.; Kaiser, S.M.; Rittmaster, R.S.; Shlossberg, A.H.; et al. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat. Genet. 2006, 38, 1248–1250. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Abou Tayoun, A.N.; Rehm, H.L. Genetic variation in the Middle East-an opportunity to advance the human genetics field. Genome Med. 2020, 12, 116. [Google Scholar] [CrossRef]
- Thauvin-Robinet, C.; Lee, J.S.; Lopez, E.; Herranz-Pérez, V.; Shida, T.; Franco, B.; Jego, L.; Ye, F.; Pasquier, L.; Loget, P.; et al. The oral-facial-digital syndrome gene C2CD3 encodes a positive regulator of centriole elongation. Nat. Genet. 2014, 46, 905–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alby, C.; Piquand, K.; Huber, C.; Megarbané, A.; Ichkou, A.; Legendre, M.; Pelluard, F.; Encha-Ravazi, F.; Abi-Tayeh, G.; Bessières, B.; et al. Mutations in KIAA0586 Cause Lethal Ciliopathies Ranging from a Hydrolethalus Phenotype to Short-Rib Polydactyly Syndrome. Am. J. Hum. Genet. 2015, 97, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoetzel, C.; Laurier, V.; Davis, E.E.; Muller, J.; Rix, S.; Badano, J.L.; Leitch, C.C.; Salem, N.; Chouery, E.; Corbani, S.; et al. BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat. Genet. 2006, 38, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Rutland, J.; de Iongh, R.U. Random ciliary orientation. A cause of respiratory tract disease. N. Eng. J. Med. 1990, 323, 1681–1684. [Google Scholar] [CrossRef] [PubMed]
- Megarbane, A.; Ghanem, I.; Waked, N.; Dagher, F. A newly recognized autosomal recessive syndrome with short stature and oculo-skeletal involvement. Am. J. Med. Genet. Part A 2006, 140, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- Singer, S.; Davidovitch, N.; Abu Fraiha, Y.; Abu Freha, N. Consanguinity and genetic diseases among the Bedouin population in the Negev. J. Community Genet. 2020, 11, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Afzal, R.M.; Lund, A.M.; Skovby, F. The impact of consanguinity on the frequency of inborn errors of metabolism. Dan. Med. J. 2018, 65, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Alkuraya, F.S. Impact of new genomic tools on the practice of clinical genetics in consanguineous populations: The Saudi experience. Clin. Genet. 2013, 84, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Khneisser, I.; Adib, S.; Assaad, S.; Megarbane, A.; Karam, P. Cost-benefit analysis: Newborn screening for inborn errors of metabolism in Lebanon. J. Med. Screen. 2015, 22, 182–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Phenotype OMIM Number | Related Gene Record | Gene/Locus OMIM Number |
|---|---|---|---|
| Diseases on CTGA not on OMIM (numbers in parentheses denote number of patients described) | |||
| Brachytelephalangy with Mental Retardation, Peculiar Face and Short Stature (1 family, 2 patients) | |||
| Congenital Contractures, Short Stature, Abnormal Face, Microcephaly, Scoliosis, Hip Dislocation, and Severe Psychomotor Retardation (2 families, 2 patients) | |||
| Craniosynostosis, Telecanthus, Scalp Hair Abnormalities, and Sensorineural Deafness (1 family, 2 patients) | |||
| Discoid Lupus Erythematosus (1 family, 4 patients) | TRAF3IP3 | 607043 | |
| Intellectual Deficiency, Unclassified (at least 147 patients) | |||
| Linear and Whorled Nevoid Hypermelanosis with Cerebral Aneurysms (1 family, 1 patient) | |||
| Marfanoid Habitus-Inguinal Hernia-Advanced Bone Age Syndrome (1 family, 2 patients) | EFEMP1 | 601548 | |
| Microcephaly, Colobomatous Micropthalmia, and mental Retardation (1 family, 2 patients) | |||
| Multiple Anomalies, Mental Retardation, Megarbane-Le Merrer-El Kallab Type (1 family, 3 patients) | |||
| Multiple Congenital Anomalies, Megarbane-Rassi Type (1 family, 1 patient) | |||
| Multiple Congenital Anomalies, Mental Retardation, Ambiguous Genitalia, Microcephaly, Seizures, and Bone Malformations (1 family, 2 patients) | |||
| Myeloproliferative Disorder, Unclassified (group of 69 patients) | JAK2 | 147796 | |
| Ptosis, Mental Retardation and 2/3 Toes Syndactyly (1 family, 2 patients) | |||
| Pure Early-Onset Dementia Without Bone Cysts (1 family, 3 patients) | TREM2 | 605086 | |
| SOX11-Related Syndrome (1 family, 1 patient) | SOX11 | 600898 | |
| Tibial and Femoral Hypoplasia with ‘Hook’ Pelvis (1 family, 1 patient) | |||
| TMTC3-Related Syndrome (1 family, 2 patients) | TMTC3 | 617218 | |
| 12q24.31 Microdeletion Syndrome (1 family, 2 patients) | |||
| 8p23.1 Microdeletion Syndrome (1 family, 1 patient) | |||
| Subacute Thyroiditis (1 family, 3 patients) | HLA-B | 142830 | |
| Chromosome 10p Duplication Syndrome (1 family, 1 patient) | |||
| Chromosome 7q Duplication Syndrome (1 family, 1 patient) | |||
| Trisomy 17p (1 family, 1 patient) | |||
| Chromosome 2p Duplication Syndrome (1 family, 1 patient) | |||
| Diseases exclusively reported in Lebanon among Arab countries | |||
| Acrodysostosis 2 With or without Hormone Resistance | 614613 | PDE4D | 600129 |
| Alveolar Soft Part Sarcoma | 606243 | ASPSCR1; TFE3 | 606236; 314310 |
| Amyotrophy, Hereditary Neuralgic | 162100 | SEPT9 | 604061 |
| Arrhythmogenic Right Ventricular Dysplasia, Familial, 11 | 610476 | DSC2 | 125645 |
| Arrhythmogenic Right ventricular Dysplasia, Familial, 9 | 609040 | PKP2 | 602861 |
| Atrial Septal Defect 4 | 611363 | TBX20 | 606061 |
| Atrial Septal Defect 5 | 612794 | ACTC1 | 102540 |
| Atrial Septal Defect, Secundum, with Various Cardiac and Noncardiac Defects * | 603642 | ||
| Bardet-Biedl Syndrome 12 | 615989 | BBS12 | 610683 |
| Benign Chronic Pemphigus | 169600 | ATP2C1 | 604384 |
| Bietti Crystalline Corneoretinal Dystrophy | 210370 | CYP4V2 | 608614 |
| Borjeson-Forssman-Lehmann Syndrome | 301900 | PHF6 | 300414 |
| Branchiogenic-Deafness Syndrome | 609166 | ||
| Brown-Vialetto-Van Laere Syndrome 2 # | 614707 | SLC52A2 | 607882 |
| Brunner Syndrome | 300615 | MAOA | 309850 |
| Cardiomyopathy, Dilated, 1KK | 615248 | MYPN | 608517 |
| Cardiomyopathy, Dilated, with Hypergonadotropic Hypogonadism | 212112 | ||
| Cataract 11, Multiple Types | 610623 | PITX3 | 602669 |
| CDAGS Syndrome | 603116 | ||
| Cerebral Creatine Deficiency Syndrome 1 | 300352 | SLC6A8 | 300036 |
| Char Syndrome | 169100 | TFAP2B | 601601 |
| Ciliary Discoordination due to Random Ciliary Orientation | 215518 | ||
| Ciliary Dyskinesia, Primary, 3 | 608644 | DNAH5 | 603335 |
| Clouston Syndrome | 129500 | ||
| Coffin-Siris Syndrome 4 | 614609 | SMARCA4 | 603254 |
| Combined Oxidative Phosphorylation Deficiency 1 # | 609060 | GFM1 | 606639 |
| Congenital Hemidysplasia with Ichthyosiform Erythroderma and Limb Defects | 308050 | ||
| Cornelia de Lange Syndrome 5 | 300882 | HDAC8 | 300269 |
| Cutis Laxa, Autosomal Recessive, Type IA | 219100 | ELN; FBLN5 | 130160; 604580 |
| Deafness, Autosomal Recessive 14 *,# | 603678 | ||
| Developmental and Epileptic Encephalopathy 13 | 614558 | SCN8A | 600702 |
| Developmental and Epileptic Encephalopathy 42 | 617106 | CACNA1A | 601011 |
| Developmental And Epileptic Encephalopathy 63 | 617976 | CPLX1 | 605032 |
| Diamond-Blackfan Anemia 6 | 612561 | RPL5 | 603634 |
| Diastrophic Dysplasia | 222600 | SLC26A2 | 606718 |
| Dislocated Elbows, Bowed Tibias, Scoliosis, Deafness, Cataract, Microcephaly, and Mental Retardation * | 603133 | ||
| Dubowitz Syndrome | 223370 | ||
| Dystonia 17, Torsion, Autosomal Recessive *,# | 612406 | ||
| Dystonia, Childhood-Onset, with Optic Atrophy and Basal Ganglia Abnormalities | 617282 | MECR | 608205 |
| Ectodermal Dysplasia and Neurosensory Deafness | 224800 | ||
| Ehlers-Danlos Syndrome, Arthrochalasia Type, 2 | 617821 | COL1A2 | 120160 |
| Ehlers-Danlos Syndrome, classic type, 1 | 130000 | COL1A1; COL5A1 | 120150; 120215 |
| Emery-Dreifuss Muscular Dystrophy 5, Autosomal Dominant | 612999 | SYNE2 | 608442 |
| Enterocolitis | 226150 | ||
| Epidermodysplasia Verruciformis, Susceptibility to, 2 | 618231 | TMC8 | 605829 |
| Epidermolysis Bullosa with Congenital Localized Absence of Skin and Deformity of Nails | 132000 | COL7A1 | 120120 |
| Epilepsy, Nocturnal Frontal Lobe, 1 | 600513 | CHRNA4 | 118504 |
| Factor XI Deficiency | 612416 | ||
| Familial Mediterranean Fever, Autosomal Dominant | 134610 | MEFV | 608107 |
| Fanconi Anemia, Complementation Group D1 | 605724 | BRCA2 | 600185 |
| Fanconi Anemia, Complementation Group E | 600901 | FANCE | 613976 |
| Fanconi Anemia, Complementation Group I | 609053 | FANCI | 611360 |
| Fanconi Anemia, Complementation group N | 610832 | PALB2 | 610355 |
| Fever, Familial Lifelong Persistent *,# | 228400 | ||
| Fibromatosis, Gingival, with Hypertrichosis and Mental Retardation | 605400 | ||
| Frontotemporal Dysplasia and/or Amyotrophic Lateral Sclerosis 4 | 616439 | TBK1 | 604834 |
| Fructose Intolerance, Hereditary | 229600 | ALDOB | 612724 |
| Generalized Epilepsy with Febrile Seizures Plus, Type 7 | 613863 | SCN9A | 603415 |
| Hymen, Imperforate | 237100 | ||
| Hyperalphalipoproteinemia 1 | 143470 | CETP | 118470 |
| Hypercarotenemia And Vitamin A Deficiency, Autosomal Recessive * | 277350 | ||
| Hyperphenylalaninemia, BH4-Deficient, B | 233910 | ||
| Hypophosphatemic Rickets, Autosomal Recessive, 1 | 241520 | DMP1 | 600980 |
| Ichthyosis, Congenital, Autosomal Recessive, 10 | 615024 | PNPLA1 | 612121 |
| Immunodeficiency 69 *,# | 618963 | IFNG | 147570 |
| Immunodeficiency with Defective T-Cell Response to Interleukin 1 * | 243110 | ||
| Infantile Liver Failure Syndrome 2 | 616483 | NBAS | 608025 |
| Inflammatory Bowel Disease 28, Autosomal Recessive # | 613148 | IL10RA | 146933 |
| Inflammatory Skin and Bowel Disease, Neonatal, 1 *,# | 614328 | ADAM17 | 603639 |
| Insulin-like Growth Factor I, Resistance to | 270450 | IGF1R | 147370 |
| Intellectual Development Disorder with Short Stature, Facial Anomalies and Speech Defects *,# | 606220 | FBXL3 | 605653 |
| Intellectual Developmental Disorder 62 | 618793 | DLG4 | 602887 |
| Internal Carotid Artery, Spontaneous Dissection of | 147820 | MTHFR | 607093 |
| Joubert Syndrome 22 | 615665 | PDE6D | 602676 |
| Koolen-De Vries Syndrome | 610443 | KANSL1 | 612452 |
| Laurin-Sandrow Syndrome | 135750 | ||
| Leber Congenital Amaurosis 7 | 613829 | CRX | 602225 |
| Lentigines | 150900 | ||
| Lethal Congenital Contracture Syndrome 7 | 616286 | CNTNAP1 | 602346 |
| Loeys-Dietz Syndrome 1 | 609192 | ||
| Loeys-Dietz Syndrome 5 | 615582 | TGFB3 | 190230 |
| Lujan-Fryns Syndrome | 309520 | ||
| Lymphoproliferative Syndrome, X-Linked, 2 | 300635 | XIAP | 300079 |
| Macrocephaly-Capillary Malformation | 602501 | ||
| Mannose 6-Phosphate Receptor Recognition Defect, Lebanese Type * | 154570 | ||
| Mental Retardation with Optic Atrophy, Facial Dysmorphism, Microcephaly, and Short Stature | 609037 | ||
| Mental Retardation-Hypotonic Facies Syndrome, X-Linked, 1 | 309580 | ATRX | 300032 |
| Mental Retardation, X-Linked, Syndromic, Christianson Type | 300243 | SLC9A6 | 300231 |
| Metaphyseal Chondrodysplasia with Cone-Shaped Epiphyses, Normal Hair, and Normal Hands | 609989 | ||
| Mitochondrial Complex I Deficiency, Nuclear Type 17 # | 618239 | NDUFAF6 | 612392 |
| Mitochondrial Complex III Deficiency, Nuclear Type 6 # | 615453 | CYC1 | 123980 |
| Mitochondrial Complex III Deficiency, Nuclear Type 7 # | 615824 | UQCC2 | 614461 |
| Mitochondrial Complex IV Deficiency, Nuclear Type 12 # | 619055 | PET100 | 614770 |
| Mitochondrial DNA Depletion Syndrome 11 # | 615084 | MGME1 | 615076 |
| Muscular Dystrophy-Dystroglycanopathy (Congenital With Impaired Intellectual Impairment), Type B, 1 | 613155 | POMT1 | 607423 |
| Muscular Dystrophy-Dystroglycanopathy (Congenital with Mental Retardation), type B, 6 | 608840 | LARGE1 | 603590 |
| Muscular Dystrophy, Limb-Girdle, Autosomal Recessive 10 | 608807 | TTN | 188840 |
| Myoclonic Epilepsy, Congenital Deafness, Macular Dystrophy, and Psychiatric Disorders | 604363 | ||
| Myofibrillar Myopathy 10 # | 619040 | SVIL | 604126 |
| Myofibrillar Myopathy 11 # | 619178 | UNC45B | 611220 |
| Myopathy, Lactic Acidosis, and Sideroblastic Anemia 2 # | 613561 | YARS2 | 610957 |
| Neurofaciodigitorenal Syndrome | 256690 | ||
| Neutrophilic Dermatosis, Acute Febrile | 608068 | MEFV | 608107 |
| Night Blindness, Congenital Stationary, Type 1E # | 614565 | GPR179 | 614515 |
| Night Blindness, Congenital Stationary, Type 1H # | 617024 | GNB3 | 139130 |
| Noonan Syndrome 4 | 610733 | SOS1 | 182530 |
| Noonan Syndrome-Like Disorder with or without Juvenile Myelomonocytic Leukemia J | 613563 | CBL | 165360 |
| Occult Macular Dystrophy | 613587 | RP1L1 | 608581 |
| Odontoonychodermal Dysplasia | 257980 | WNT10A | 606268 |
| Orofaciodigital Syndrome, Type IV | 258860 | ||
| Osteogenesis Imperfecta, Type XVI # | 616229 | CREB3L1 | 616215 |
| Otopalatodigital Syndrome, Type I | 311300 | FLNA | 300017 |
| Paget Disease of bone 2, Early-onset | 602080 | ||
| Pallister-Hall Syndrome | 146510 | ||
| Parkinson Disease 7, Autosomal Recessive Early-Onset | 606324 | ||
| Pentosuria | 260800 | ||
| Peripheral Neuropathy, Autosomal Recessive, with or without Impaired Intellectual Development | 618124 | MCM3AP | 603294 |
| Pigmentary Disorder, Reticulate, with Systemic Manifestations | 301220 | ||
| Pitt-Hopkins Syndrome | 610954 | ||
| Premature Ovarian Failure 2B * # | 300604 | ||
| Progressive Familial Heart Block, Type IB # | 604559 | TRPM4 | 606936 |
| Pseudoachondroplasia | 177170 | COMP | 600310 |
| Ramon Syndrome | 266270 | ELMO2 | 606421 |
| Retinopathy, Pigmentary, and Mental Retardation | 268050 | VPS13A | 605978 |
| Rhizomelic Dysplasia, Scoliosis, and Retinitis Pigmentosa * | 610319 | ||
| Roifman Syndrome | 616651 | RNU4ATAC | 601428 |
| Short Stature and Facioauriculothoracic Malformations * | 609654 | ||
| Short-Rib Thoracic Dysplasia 14 With Polydactyly # | 616546 | KIAA0586 | 610178 |
| Silver-Russell Syndrome | 180860 | ||
| Skeletal Dysplasia, Rhizomelic, with Retinitis Pigmentosa * | 609047 | ||
| Spasticity, Childhood-Onset, with Hyperglycinemia # | 616859 | GLRX5 | 609588 |
| Spinal Muscular Atrophy, Distal, Autosomal Recessive # | 607088 | VRK1 | 602168 |
| Spinocerebellar Ataxia 13 | 605259 | KCNC3 | 176264 |
| Spinocerebellar Ataxia 35 | 613908 | TGM6 | 613900 |
| Spinocerebellar Ataxia, Autosomal Recessive 2 | 213200 | PMPCA | 613036 |
| Spinocerebellar Ataxia, Autosomal Recessive 24 | 617133 | UBA5 | 610552 |
| Spinocerebellar Degeneration and Corneal Dystrophy | 271310 | ||
| Spondylocostal Dysostosis 3, Autosomal Recessive # | 609813 | LFNG | 602576 |
| Chromosome 10q26 Deletion Syndrome | 609813 | ||
| Spondylocostal Dysostosis, Autosomal Recessive 2 # | 608681 | MESP2 | 605195 |
| Spondyloepimetaphyseal Dysplasia, Maroteaux Type # | 184095 | TRPV4 | 605427 |
| Spondylometaphyseal Dysplasia, Megarbane-Dagher-Melki Type # | 613320 | PAM16 | 614336 |
| Stocco Dos Santos X-Linked Mental Retardation | 300434 | SHROOM4 | 300579 |
| Surfactant Metabolism Dysfunction, Pulmonary, 1 | 265120 | SMDP1 | 265120 |
| Teeth, Supernumerary | 187100 | ||
| Testes, Rudimentary | 273150 | ||
| Thiopurines, Poor Metabolism of, 1 | 610460 | TPMT | 187680 |
| Tricuspid atresia | 605067 | NFATC1 | 600489 |
| Tubulointerstitial Kidney Disease, Autosomal Dominant, 2 | 174000 | MUC1 | 158340 |
| Ulnar Hypoplasia | 191440 | ||
| Variegate Porphyria | 176200 | PPOX | 600923 |
| Vertebral, Cardiac, Renal, and Limb Defects Syndrome 2 # | 617661 | KYNU | 605197 |
| Vibratory Urticaria | 125630 | ADGRE2 | 606100 |
| Vitamin K-Dependent Clotting Factors, Combined Deficiency of, 2 # | 607473 | VKORC1 | 608547 |
| Diseases first linked or mapped in Lebanese subjects | |||
| Alpha/Beta T-Cell Lymphoma with Gamma/Delta T-Cell Expansion, Severe Cytomegalovirus Infection, and Autoimmunity # | 609889 | RAG1 | 179615 |
| Bardet-Biedl Syndrome 10 # | 615987 | BBS10 | 610148 |
| Charcot-Marie-Tooth Disease, Demyelinating, Type 4F # | 614895 | PRX | 605725 |
| Charcot-Marie-Tooth Disease, Type 4H # | 609311 | FGD4 | 611104 |
| Deafness, Autosomal Recessive 13 # | 603098 | ||
| Deafness, Autosomal Recessive 21 # | 603629 | TECTA | 602574 |
| Deafness, Autosomal Recessive 9 # | 601071 | OTOF | 603681 |
| Dihydropyrimidinase Deficiency # | 222748 | DPYS | 613326 |
| Galloway-Mowat Syndrome 1 # | 251300 | WDR73; ZNF592 | 616144; 613624 |
| Hepatic Venoocclusive Disease with Immunodeficiency # | 235550 | SP110 | 604457 |
| Hydatidiform Mole, Recurrent, 1 # | 231090 | NLRP7 | 609661 |
| Hypercholesterolemia, Familial, 4 # | 603813 | LDLRAP1 | 605747 |
| Ichthyosis, Congenital, Autosomal Recessive 13 # | 617574 | ||
| Immunodeficiency 12 # | 615468 | ||
| Immunodeficiency 40 # | 616433 | DOCK2 | 603122 |
| Immunodeficiency 56 # | 615207 | IL21R | 605383 |
| Lipodystrophy, Congenital Generalized, Type 2 # | 269700 | BSCL2 | 606158 |
| Pancreatic Agenesis 2 # | 615935 | PTF1A | 607194 |
| Spinal Muscular Atrophy, Distal, Autosomal Recessive, 1 # | 604320 | IGHMBP2; REEP1 | 600502; 609139 |
| Weill-Marchesani Syndrome, Autosomal Recessive # | 277600 | ADAMTS10 | 608990 |
| Microphthalmia with Limb Anomalies # | 206920 | FNBP4; SMOC1 | 615265; 608488 |
| Geleophysic Dysplasia 1 # | 231050 | FBN1 | 134797 |
| Frank-Ter Haar Syndrome # | 249420 | SH3PXD2B | 613293 |
| Orofaciodigital Syndrome XIV # | 615948 | C2CD3 | 615944 |
| Baller-Gerold Syndrome # | 218600 | RECQL4 | 603780 |
| Individual Subject Entries | Group Subject Entries |
|---|---|
| Intellectual disability | Diabetes |
| Hearing impairment | Coronary Artery Disease |
| Global developmental delay | Abnormal cholesterol levels |
| Seizures | Familial Mediterranean Fever |
| Short stature | Anemia |
| Muscular hypotonia | Inflammatory bowel disease |
| Delayed speech and language development | Hypertension |
| Failure to thrive | Obesity |
| Periodic fever | Myeloproliferative disorder |
| Atrial septal defect | Myocardial Infarction |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bizzari, S.; Nair, P.; Deepthi, A.; Hana, S.; Al-Ali, M.T.; Megarbané, A.; El-Hayek, S. Catalogue for Transmission Genetics in Arabs (CTGA) Database: Analysing Lebanese Data on Genetic Disorders. Genes 2021, 12, 1518. https://doi.org/10.3390/genes12101518
Bizzari S, Nair P, Deepthi A, Hana S, Al-Ali MT, Megarbané A, El-Hayek S. Catalogue for Transmission Genetics in Arabs (CTGA) Database: Analysing Lebanese Data on Genetic Disorders. Genes. 2021; 12(10):1518. https://doi.org/10.3390/genes12101518
Chicago/Turabian StyleBizzari, Sami, Pratibha Nair, Asha Deepthi, Sayeeda Hana, Mahmoud Taleb Al-Ali, André Megarbané, and Stephany El-Hayek. 2021. "Catalogue for Transmission Genetics in Arabs (CTGA) Database: Analysing Lebanese Data on Genetic Disorders" Genes 12, no. 10: 1518. https://doi.org/10.3390/genes12101518
APA StyleBizzari, S., Nair, P., Deepthi, A., Hana, S., Al-Ali, M. T., Megarbané, A., & El-Hayek, S. (2021). Catalogue for Transmission Genetics in Arabs (CTGA) Database: Analysing Lebanese Data on Genetic Disorders. Genes, 12(10), 1518. https://doi.org/10.3390/genes12101518