
Characterization of a Missense Mutation in the Catalytic Domain and a Splicing Mutation of Coagulation Factor X Compound Heterozygous in a Chinese Pedigree


Abstract
:1. Introduction
2. Materials and Methods
2.1. Nomenclature
2.2. Patients and Samples
2.3. Routine Coagulation and Coagulation Factor Assays
2.4. Gene Analysis
2.5. Construction of Plasmid Expression Vectors
2.6. Transfection
2.7. Purification of Recombinant FX-WT and FX-Ile265Thr
2.8. Activation of Recombinant FX
2.9. Clotting Activity
2.10. Thrombin Generation Assay
2.11. Minigene Assay
2.12. Bioinformatics Analysis
3. Results
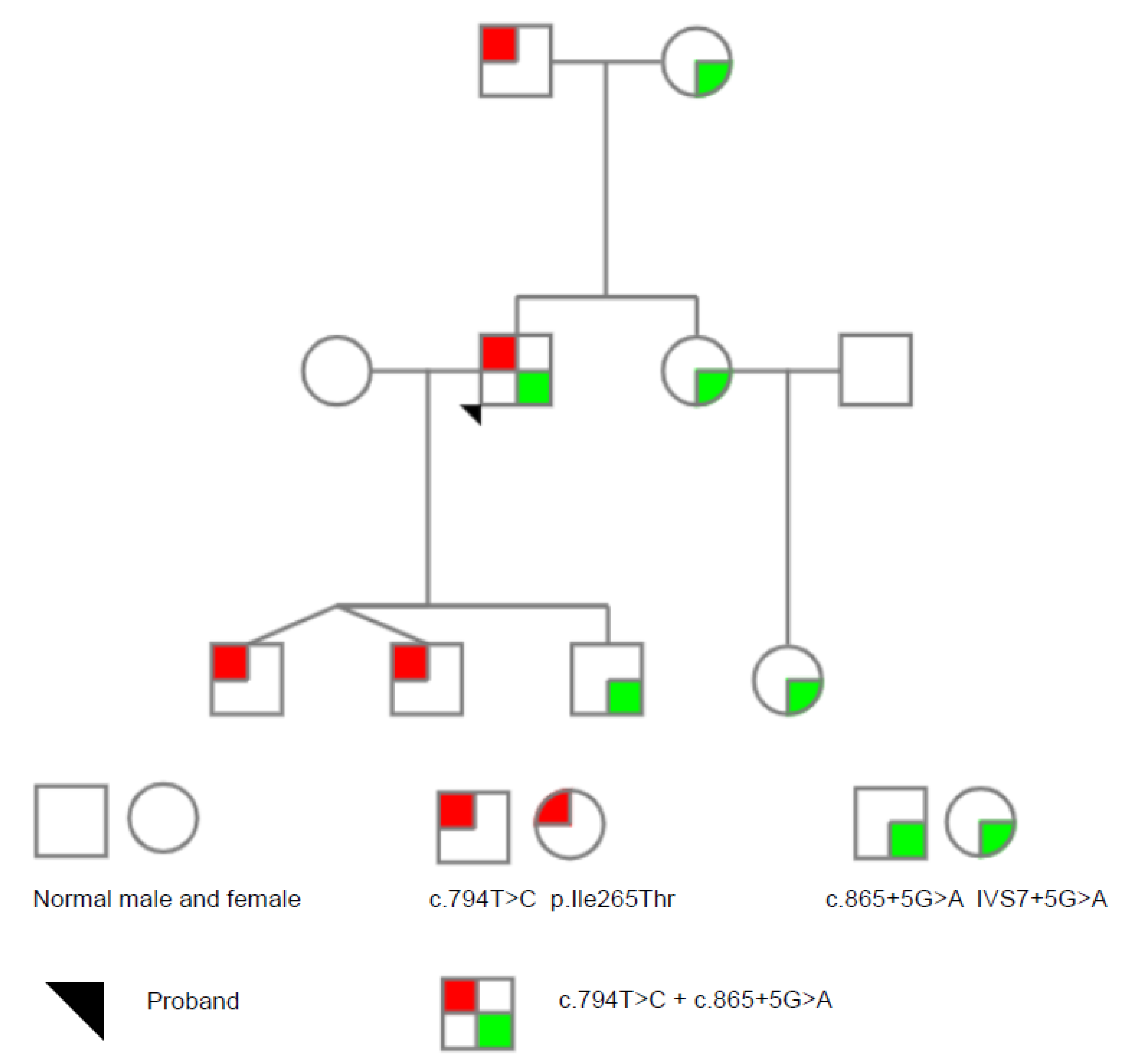
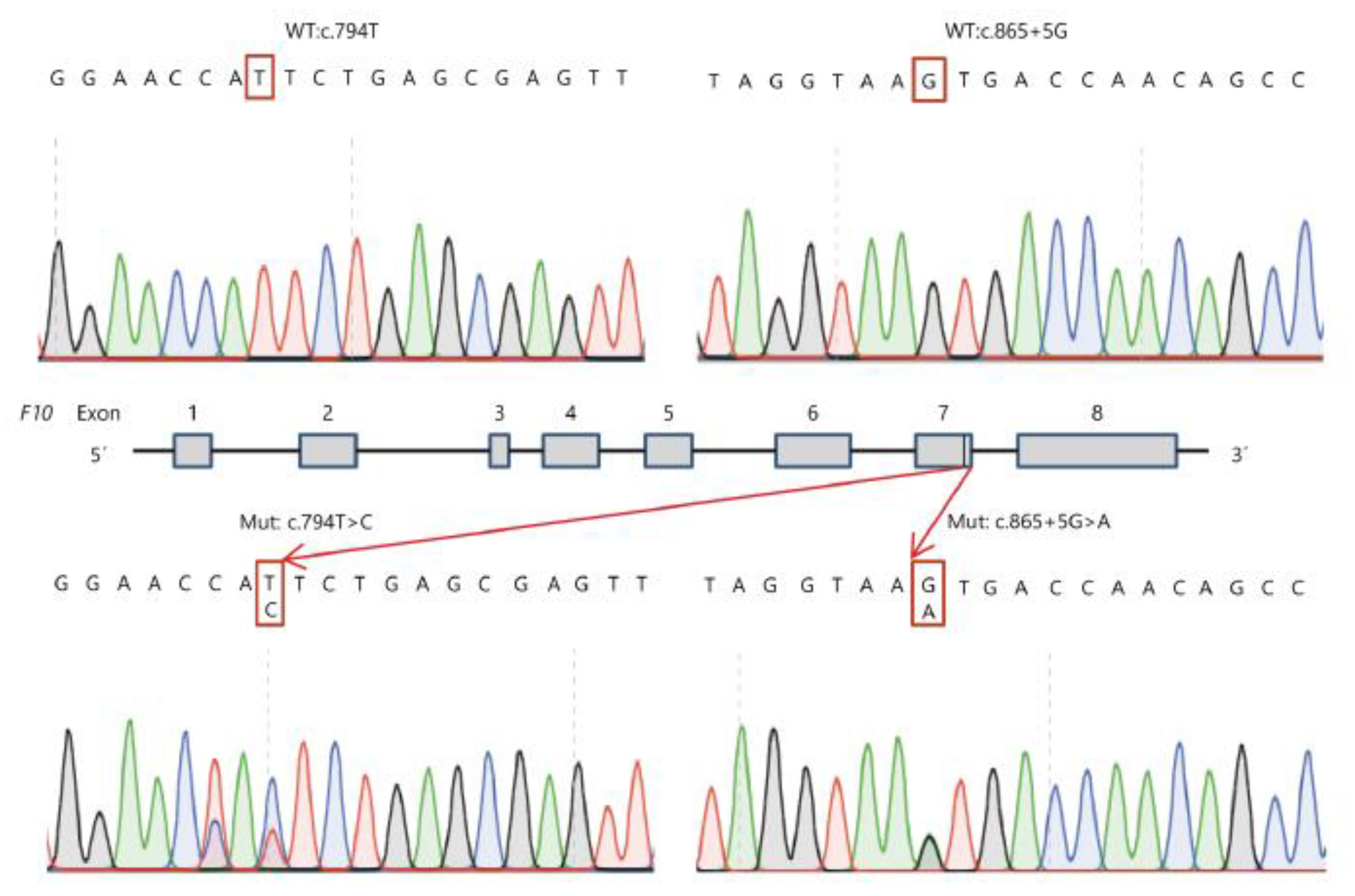
3.1. Genetic Analysis
3.2. Routine Coagulation Test and Coagulation Factors
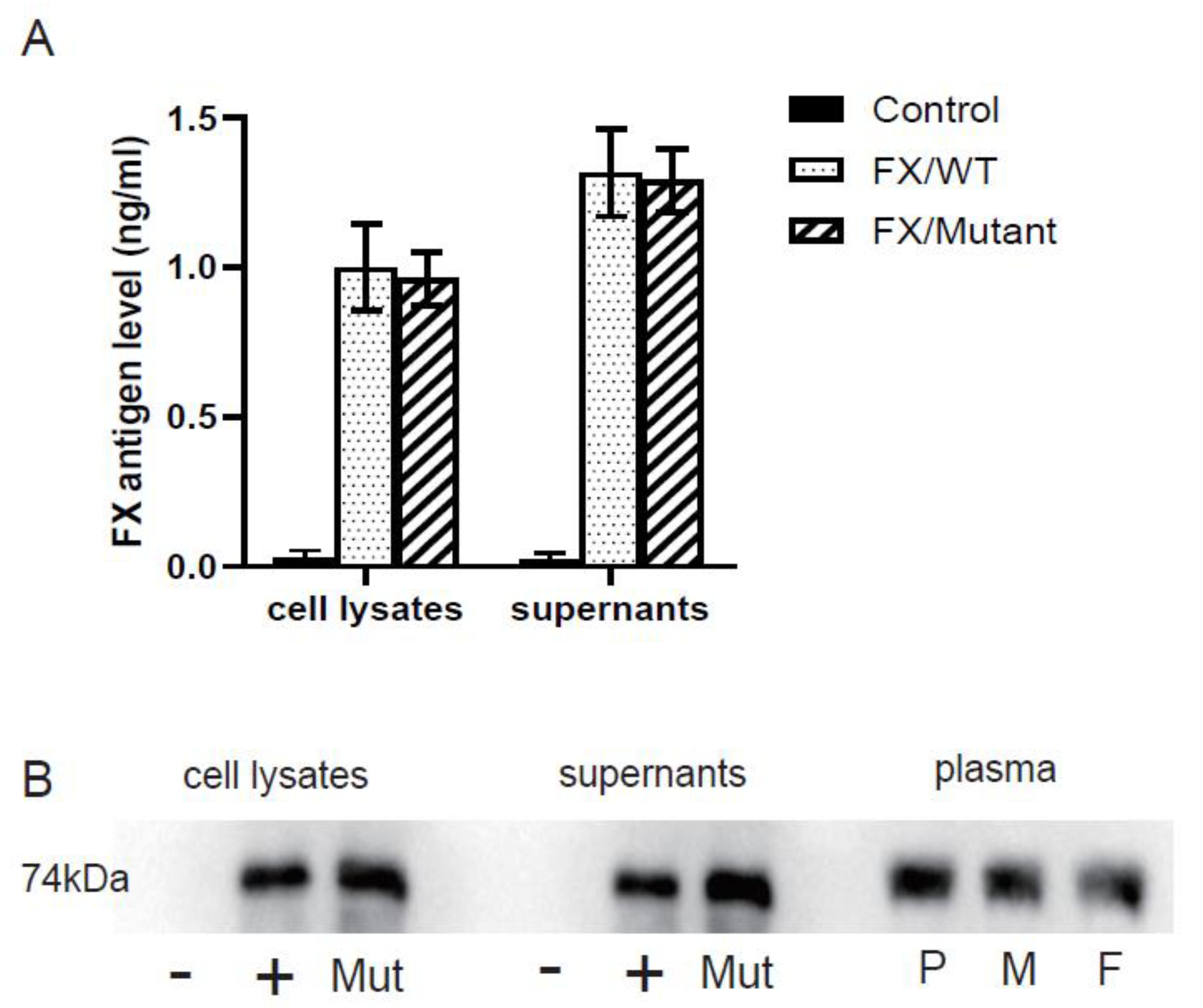
3.3. In-Vitro Expression
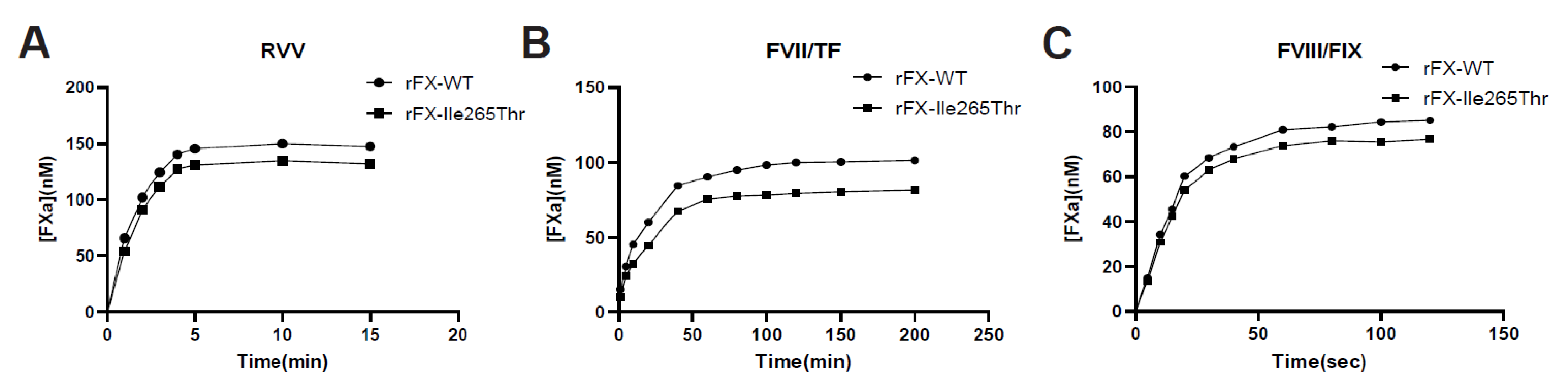
3.4. Activation of Recombinant FX-WT and FX-Ile265Thr
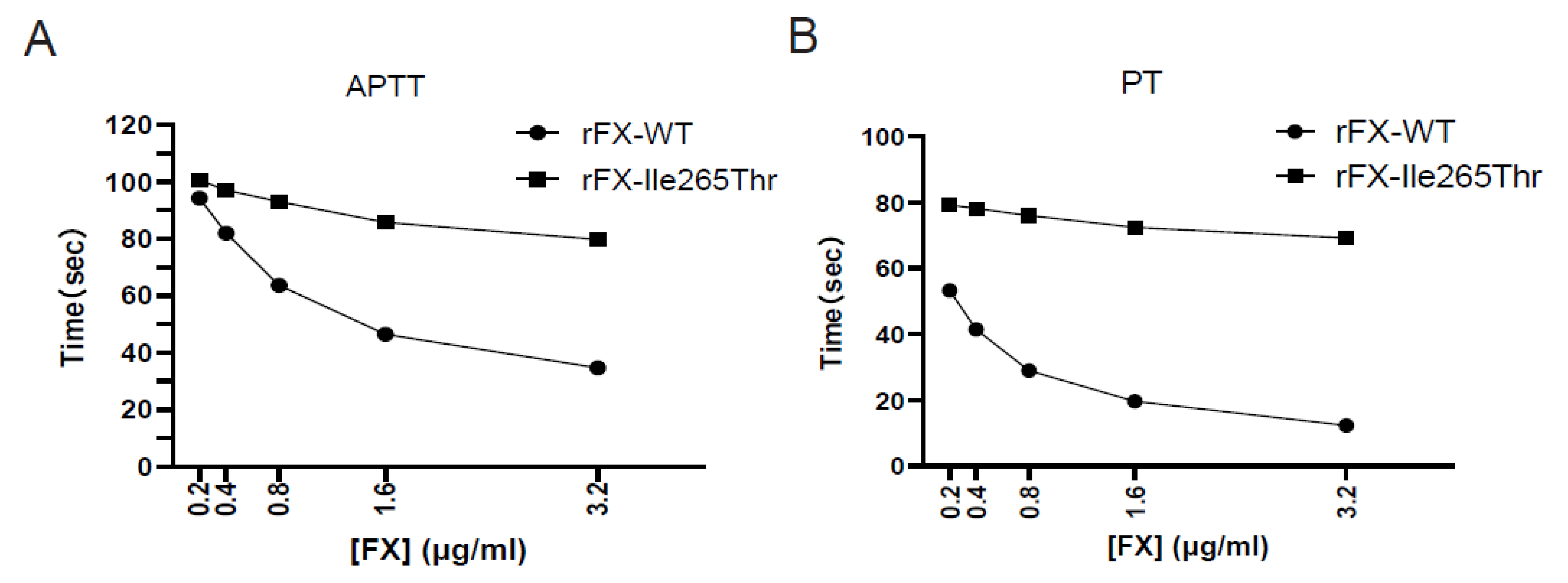
3.5. Clotting Activity of Recombinant FX
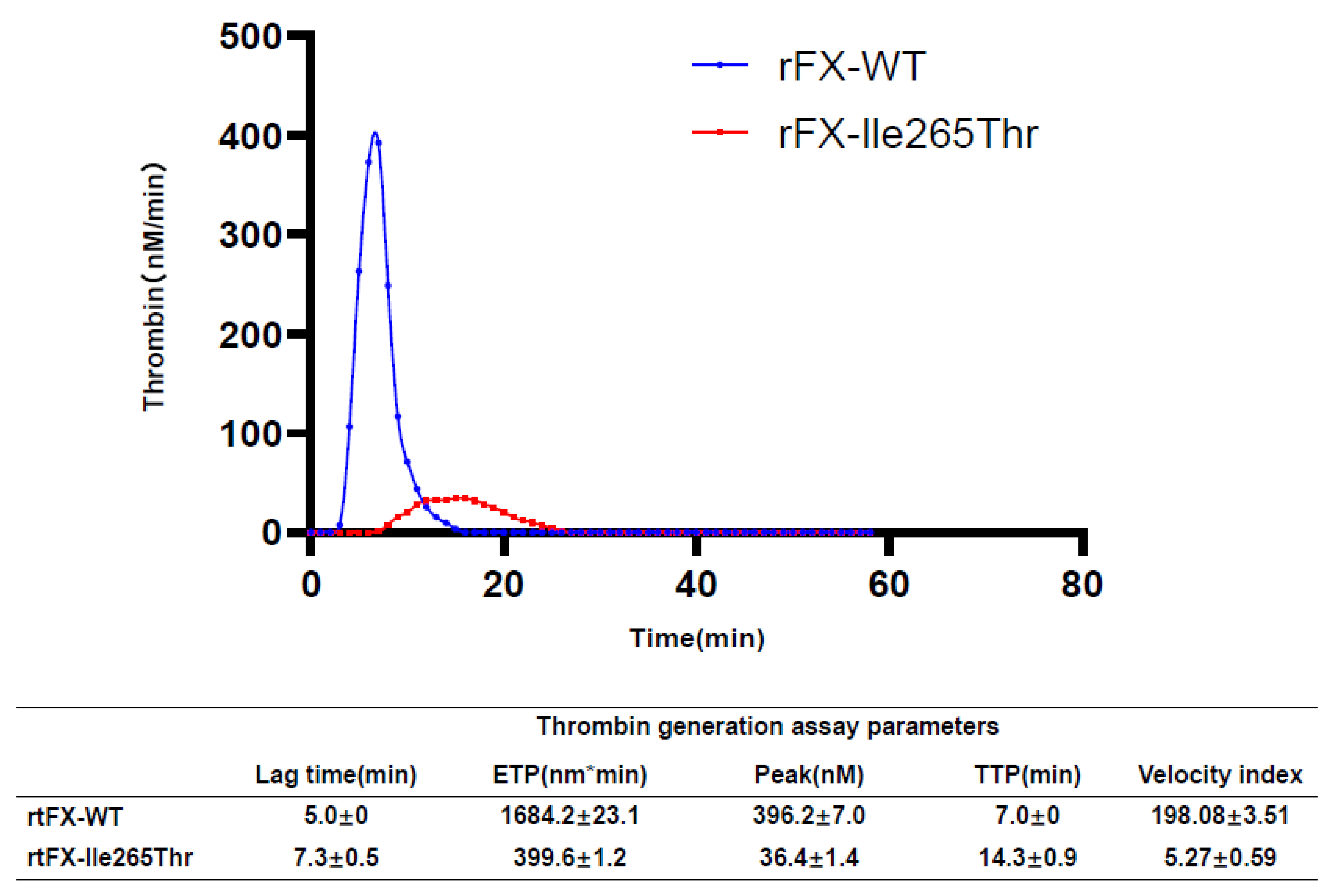
3.6. Thrombin Generation Assay Analysis
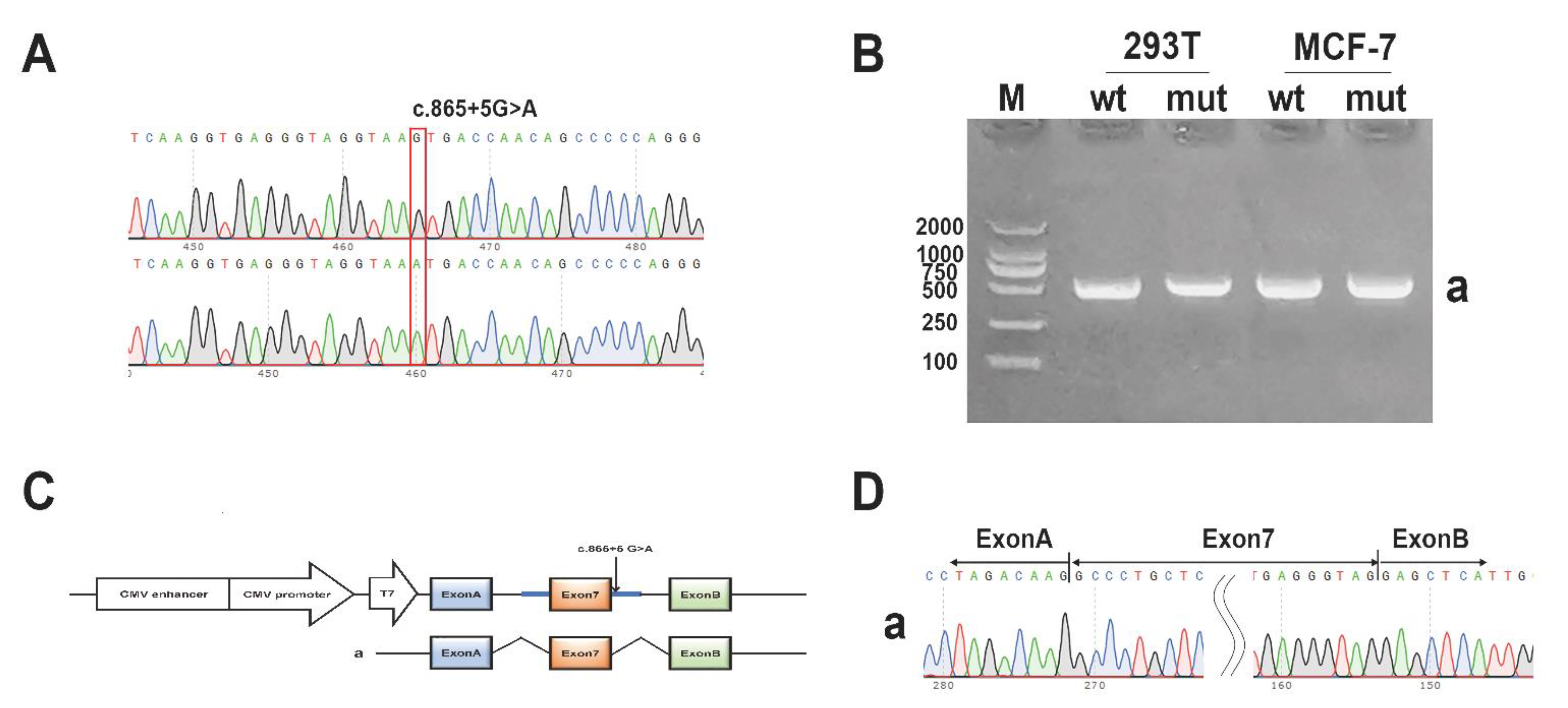
3.7. Minigene Assay
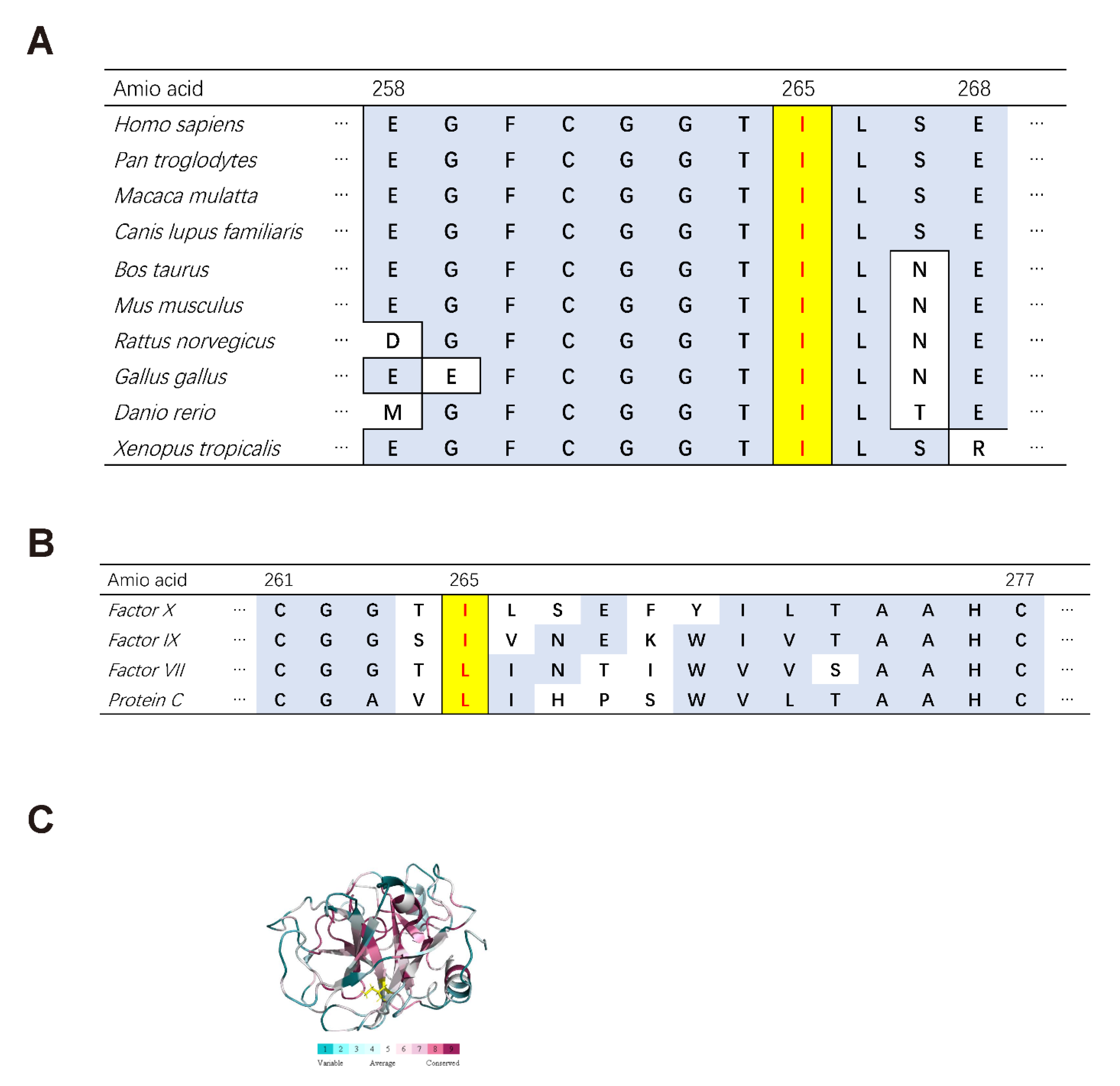
3.8. Bioinformatics Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Transparency Statement
References
- Bajaj, S.P.; Mann, K.G. Simultaneous purification of bovine prothrombin and factor X. Activation of prothrombin by trypsin-activated factor X. J. Biol. Chem. 1973, 248, 7729–7741. [Google Scholar] [CrossRef]
- Kisiel, W.; Hermodson, M.A.; Davie, E.W. Factor X activating enzyme from Russell's viper venom: Isolation and characterization. Biochem.-Us 1976, 15, 4901–4906. [Google Scholar] [CrossRef]
- Rao, L.V.; Rapaport, S.I. Activation of factor VII bound to tissue factor: A key early step in the tissue factor pathway of blood coagulation. Proc. Natl. Acad. Sci. USA 1988, 85, 6687–6691. [Google Scholar] [CrossRef] [PubMed]
- Furie, B.; Furie, B.C. The molecular basis of blood coagulation. Cell 1988, 53, 505–518. [Google Scholar] [CrossRef]
- Dewerchin, M.; Liang, Z.; Moons, L.; Carmeliet, P.; Castellino, F.J.; Collen, D.; Rosen, E.D. Blood coagulation factor x deficiency causes partial embryonic lethality and fatal neonatal bleeding in mice. Thromb. Haemost. 2000, 83, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Peyvandi, F.; Mannucci, P.M.; Lak, M.; Abdoullahi, M.; Zeinali, S.; Sharifian, R.; Perry, D. Congenital factor X deficiency: Spectrum of bleeding symptoms in 32 Iranian patients. Brit. J. Haematol. 1998, 102, 626–628. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, F.H.; Auerswald, G.; Ruiz-Saez, A.; Navarrete, M.; Pollmann, H.; Lopaciuk, S.; Batorova, A.; Wulff, K. Factor X deficiency: Clinical manifestation of 102 subjects from Europe and Latin America with mutations in the factor 10 gene. Haemophilia 2006, 12, 479–489. [Google Scholar] [CrossRef]
- Karimi, M.; Menegatti, M.; Afrasiabi, A.; Sarikhani, S.; Peyvandi, F. Phenotype and genotype report on homozygous and heterozygous patients with congenital factor X deficiency. Haematologica 2008, 93, 934–938. [Google Scholar] [CrossRef]
- Peyvandi, F.; Menegatti, M.; Santagostino, E.; Akhavan, S.; Uprichard, J.; Perry, D.J.; Perkins, S.J.; Mannucci, P.M. Gene mutations and three-dimensional structural analysis in 13 families with severe factor X deficiency. Brit. J. Haematol. 2002, 117, 685–692. [Google Scholar] [CrossRef]
- Menegatti, M.; Peyvandi, F. Treatment of rare factor deficiencies other than hemophilia. Blood 2019, 133, 415–424. [Google Scholar] [CrossRef]
- Jayandharan, G.; Viswabandya, A.; Baidya, S.; Nair, S.C.; Shaji, R.V.; George, B.; Chandy, M.; Srivastava, A. Six novel mutations including triple heterozygosity for Phe31Ser, 514delT and 516T-->G factor X gene mutations are responsible for congenital factor X deficiency in patients of Nepali and Indian origin. J. Thromb. Haemost. 2005, 3, 1482–1487. [Google Scholar] [CrossRef]
- Mannucci, P.M.; Duga, S.; Peyvandi, F. Recessively inherited coagulation disorders. Blood 2004, 104, 1243–1252. [Google Scholar] [CrossRef]
- Wang, W.B.; Fu, Q.H.; Zhou, R.F.; Wu, W.M.; Ding, Q.L.; Hu, Y.Q.; Wang, X.F.; Wang, H.L.; Wang, Z.Y. Molecular characterization of two novel mutations causing factor X deficiency in a Chinese pedigree. Haemophilia 2005, 11, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Chen, Q.; Ding, Q.; Wu, F.; Wang, X.; Xi, X.; Wang, H. Six novel missense mutations causing factor X deficiency and application of thrombin generation test. Thromb. Res. 2013, 131, 554–559. [Google Scholar] [CrossRef]
- Ding, Q.; Shen, Y.; Yang, L.; Wang, X.; Rezaie, A.R. The missense Thr211Pro mutation in the factor X activation peptide of a bleeding patient causes molecular defect in the clotting cascade. Thromb. Haemost. 2013, 110, 53–61. [Google Scholar]
- Zhou, J.W.; Liang, Q.; Chen, Q.; Xie, Y.; Ding, Q.L.; Wang, X.F.; Xi, X.D.; Wang, H.L. Molecular defects in the factor X gene caused by novel heterozygous mutations IVS5+1G>A and Asp409del. Haemophilia 2013, 19, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Yang, L.; Manithody, C.; Wang, X.; Rezaie, A.R. Molecular basis of the clotting defect in a bleeding patient missing the Asp-185 codon in the factor X gene. Thromb. Res. 2014, 134, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ma, J.; Liu, X.; Wang, Y.; Wang, H.; Wang, L.; Ding, Q.; Chu, X.; Hou, M. A novel factor X gene mutation Val (GTC) 384Ala (GCC) in a Chinese family resulting in congenital factor X deficiency. Int. J. Clin. Exp. Med. 2015, 8, 10095–10098. [Google Scholar]
- Jin, Y.; Hao, X.; Cheng, X.; Yang, L.; Chen, Y.; Xie, H.; Wang, Y.; Wang, M. [Homozygous missense mutation p.Val298Met of F10 gene causing hereditary coagulation factor X deficiency in a Chinese pedigree]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2016, 33, 296–299. [Google Scholar] [PubMed]
- Jin, Y.; Cheng, X.; Zheng, J.; Xia, H.; Yang, L.; Wang, M. A novel factor X mutation Cys81 by Arg and a reported factor VII polymorphism Arg353 replaced by Gln co-occured in a patient. Blood Coagul. Fibrinolysis 2018, 29, 67–74. [Google Scholar] [CrossRef]
- Li, F.; Chen, C.; Qu, S.; Zhao, M.; Xie, X.; Wu, X.; Li, L.; Wang, X.; Ding, Q.; Xu, Q.; et al. The Disulfide Bond between Cys22 and Cys27 in the Protease Domain Modulate Clotting Activity of Coagulation Factor X. Thromb. Haemost. 2019, 119, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Lin, W.; Ji, H.; Su, M.; Zhao, X.; Wang, C. A Compound Heterozygosis of Two Novel Mutations Causes Factor X Deficiency in a Chinese Pedigree. Acta Haematol. 2021, 144, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, K.; Wang, G.; Zhang, C.; Zhao, B.; Liu, X.; Qin, X.; Yang, L. Molecular mechanism of a novel Ser362Asn mutation causing inherited FX deficiency in a Chinese family. Int. J. Hematol. 2020, 112, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shi, L.; Zhang, K.; Zhang, Y.; Hu, S.; Zhao, T.; Teng, H.; Li, X.; Jiang, Y.; Ji, L.; et al. VarCards: An integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 2018, 46, D1039–D1048. [Google Scholar] [CrossRef]
- Mumford, A.D.; Ackroyd, S.; Alikhan, R.; Bowles, L.; Chowdary, P.; Grainger, J.; Mainwaring, J.; Mathias, M.; O’Connell, N. Guideline for the diagnosis and management of the rare coagulation disorders: A United Kingdom Haemophilia Centre Doctors’ Organization guideline on behalf of the British Committee for Standards in Haematology. Br. J. Haematol. 2014, 167, 304–326. [Google Scholar] [CrossRef]
- Karimi, M.; Vafafar, A.; Haghpanah, S.; Payandeh, M.; Eshghi, P.; Hoofar, H.; Afrasiabi, A.; Gerdabi, J.; Ardeshiri, R.; Menegatti, M.; et al. Efficacy of prophylaxis and genotype-phenotype correlation in patients with severe Factor X deficiency in Iran. Haemophilia 2012, 18, 211–215. [Google Scholar] [CrossRef]
- Escobar, M.A.; Auerswald, G.; Austin, S.; Huang, J.N.; Norton, M.; Millar, C.M. Experience of a new high-purity factor X concentrate in subjects with hereditary factor X deficiency undergoing surgery. Haemophilia 2016, 22, 713–720. [Google Scholar] [CrossRef]
- Nagaya, S.; Akiyama, M.; Murakami, M.; Sekiya, A.; Asakura, H.; Morishita, E. Congenital coagulation factor X deficiency: Genetic analysis of five patients and functional characterization of mutant factor X proteins. Haemophilia 2018, 24, 774–785. [Google Scholar] [CrossRef]
- Millar, D.S.; Elliston, L.; Deex, P.; Krawczak, M.; Wacey, A.I.; Reynaud, J.; Nieuwenhuis, H.K.; Bolton-Maggs, P.; Mannucci, P.M.; Reverter, J.C.; et al. Molecular analysis of the genotype-phenotype relationship in factor X deficiency. Hum. Genet. 2000, 106, 249–257. [Google Scholar]
- Gaildrat, P.; Killian, A.; Martins, A.; Tournier, I.; Frebourg, T.; Tosi, M. Use of splicing reporter minigene assay to evaluate the effect on splicing of unclassified genetic variants. Methods Mol. Biol. 2010, 653, 249–257. [Google Scholar]
- Ding, Q.; Wu, W.; Fu, Q.; Wang, X.; Hu, Y.; Wang, H.; Wang, Z. Novel aberrant splicings caused by a splice site mutation (IVS1a+5g>a) in F7 gene. Thromb. Haemost. 2005, 93, 1077–1081. [Google Scholar] [PubMed]
- Giansily-Blaizot, M.; Aguilar-Martinez, P.; Biron-Andreani, C.; Jeanjean, P.; Igual, H.; Schved, J.F. Analysis of the genotypes and phenotypes of 37 unrelated patients with inherited factor VII deficiency. Eur. J. Hum. Genet. 2001, 9, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Wulff, K.; Herrmann, F.H. Twenty two novel mutations of the factor VII gene in factor VII deficiency. Hum. Mutat. 2000, 15, 489–496. [Google Scholar] [CrossRef]
- Bernardi, F.; Patracchini, P.; Gemmati, D.; Ferrati, M.; Arcieri, P.; Papacchini, M.; Redaelli, R.; Baudo, F.; Mariani, G.; Marchetti, G. Molecular analysis of factor VII deficiency in Italy: A frequent mutation (FVII Lazio) in a repeated intronic region. Hum. Genet. 1993, 92, 446–450. [Google Scholar] [CrossRef]
- Reitsma, P.H.; Poort, S.R.; Allaart, C.F.; Briëf, E.; Bertina, R.M. The Spectrum of Genetic Defects in a Panel of 40 Dutch Families with Symptomatic Protein C Deficiency Type I: Heterogeneity and Founder Effects. Blood 1991, 78, 890–894. [Google Scholar] [CrossRef]
- Balestra, D.; Maestri, I.; Branchini, A.; Ferrarese, M.; Bernardi, F.; Pinotti, M. An Altered Splicing Registry Explains the Differential ExSpeU1-Mediated Rescue of Splicing Mutations Causing Haemophilia A. Front. Genet. 2019, 10, 974. [Google Scholar] [CrossRef]
- Lombardi, S.; Leo, G.; Merlin, S.; Follenzi, A.; McVey, J.H.; Maestri, I.; Bernardi, F.; Pinotti, M.; Balestra, D. Dissection of pleiotropic effects of variants in and adjacent to F8 exon 19 and rescue of mRNA splicing and protein function. Am. J. Hum. Genet. 2021, 108, 1512–1525. [Google Scholar] [CrossRef]
- Ingerslev, J.; Herlin, T.; Sorensen, B.; Clausen, N.; Chu, K.C.; High, K.A. Severe factor X deficiency in a pair of siblings: Clinical presentation, phenotypic and genotypic features, prenatal diagnosis and treatment. Haemophilia 2007, 13, 334–336. [Google Scholar] [CrossRef]
- Deshpande, R.; Ghosh, K.; Shetty, S. Synergistic effect of factor VII gene polymorphisms causing mild factor VII deficiency in a case of severe factor X deficiency. Blood Coagul. Fibrinolysis 2017, 28, 105–106. [Google Scholar] [CrossRef]
- Borhany, M.; Buthiau, D.; Rousseau, F.; Guillot, O.; Naveena, F.; Abid, M.; Shamsi, T.; Giansily-Blaizot, M. Genotyping of five Pakistani patients with severe inherited factor X deficiency: Identification of two novel mutations. Blood Coagul. Fibrinolysis 2018, 29, 622–625. [Google Scholar] [CrossRef]
- Mitchell, M.; Gattens, M.; Kavakli, K.; Liesner, R.; Payne, J.; Norton, M.; Austin, S. Genotype analysis and identification of novel mutations in a multicentre cohort of patients with hereditary factor X deficiency. Blood Coagul. Fibrinolysis 2019, 30, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Togashi, T.; Nagaya, S.; Nagasawa, M.; Meguro-Horike, M.; Nogami, K.; Imai, Y.; Kuzasa, K.; Sekiya, A.; Horike, S.I.; Asakura, H.; et al. Genetic analysis of a compound heterozygous patient with congenital factor X deficiency and regular replacement therapy with a prothrombin complex concentrate. Int. J. Hematol. 2020, 111, 51–56. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family Members | Sex | Age (Years) | aPTT (s) | PT (s) | FX Activity % | FX Antigen % |
|---|---|---|---|---|---|---|
| Proband | M | 28 | 65.3 | 80.5 | <1 | 49.7 |
| Mother | F | 56 | 25.3 | 12.2 | 50.3 | 43.2 |
| Father | M | 58 | 26.4 | 11.3 | 57.2 | 72.0 |
| Sister | F | 32 | 30.0 | 12.7 | 40.7 | 45.3 |
| Wife | F | 26 | 24.3 | 12.3 | 93.0 | 112.2 |
| Son1 | M | 4 | 29.8 | 12.4 | 47.8 | 42.3 |
| Son2(twin) | M | 1 month | 38.2 | 12.7 | - | - |
| Son3(twin) | M | 1 month | 37.6 | 13.1 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, Y.; Ma, J.; Tang, L.V.; Lin, W.; Tao, Y.; Cheng, Z.; Hu, Y. Characterization of a Missense Mutation in the Catalytic Domain and a Splicing Mutation of Coagulation Factor X Compound Heterozygous in a Chinese Pedigree. Genes 2021, 12, 1521. https://doi.org/10.3390/genes12101521
Feng Y, Ma J, Tang LV, Lin W, Tao Y, Cheng Z, Hu Y. Characterization of a Missense Mutation in the Catalytic Domain and a Splicing Mutation of Coagulation Factor X Compound Heterozygous in a Chinese Pedigree. Genes. 2021; 12(10):1521. https://doi.org/10.3390/genes12101521
Chicago/Turabian StyleFeng, Yuanzheng, Jiewen Ma, Liang V Tang, Wenyi Lin, Yanyi Tao, Zhipeng Cheng, and Yu Hu. 2021. "Characterization of a Missense Mutation in the Catalytic Domain and a Splicing Mutation of Coagulation Factor X Compound Heterozygous in a Chinese Pedigree" Genes 12, no. 10: 1521. https://doi.org/10.3390/genes12101521
APA StyleFeng, Y., Ma, J., Tang, L. V., Lin, W., Tao, Y., Cheng, Z., & Hu, Y. (2021). Characterization of a Missense Mutation in the Catalytic Domain and a Splicing Mutation of Coagulation Factor X Compound Heterozygous in a Chinese Pedigree. Genes, 12(10), 1521. https://doi.org/10.3390/genes12101521