
The Distribution of Campylobacter jejuni Virulence Genes in Genomes Worldwide Derived from the NCBI Pathogen Detection Database

 ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Multilocus Sequence Type (MLST) Identification
2.3. In Silico Virulome Determination
2.4. Statistical Analyses
3. Results
3.1. Metadata Overview
3.2. Multilocus Sequence Type (MLST)
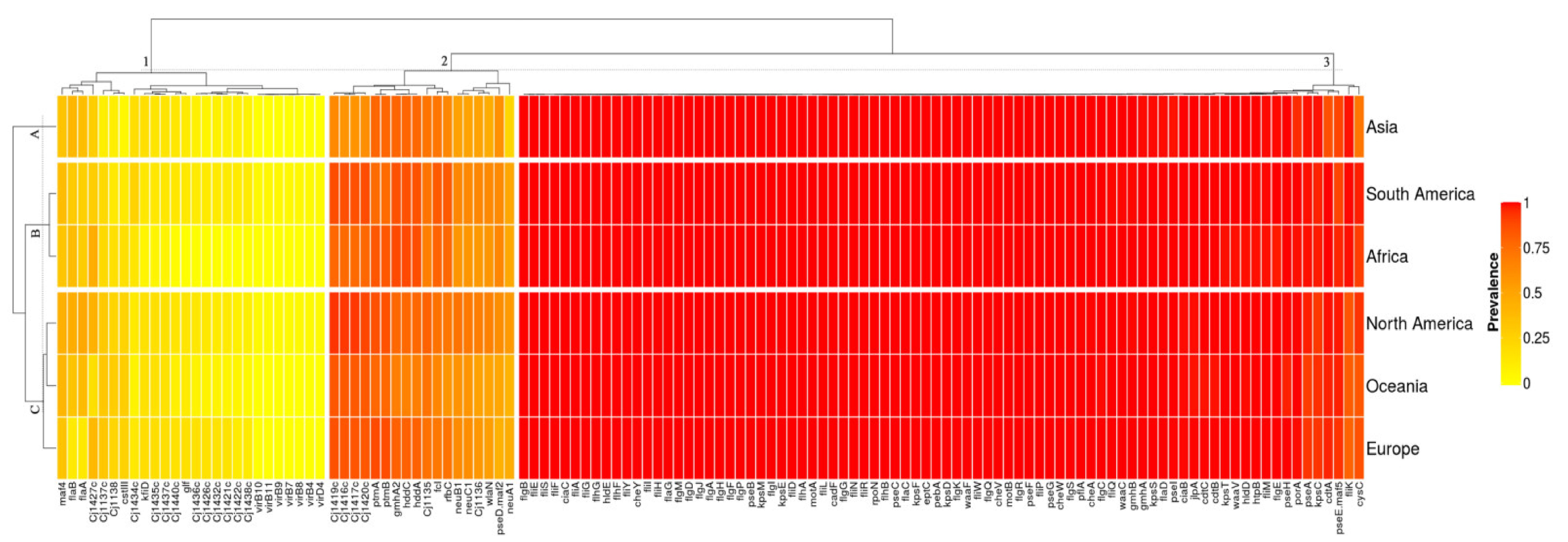
3.3. Campylobacter Jejuni Virulome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Young, K.T.; Davis, L.M.; DiRita, V.J. Campylobacter jejuni: Molecular Biology and Pathogenesis. Nat. Rev. Microbiol. 2007, 5, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Epping, L.; Walther, B.; Piro, R.M.; Knüver, M.-T.; Huber, C.; Thürmer, A.; Flieger, A.; Fruth, A.; Janecko, N.; Wieler, L.H.; et al. Genome-Wide Insights into Population Structure and Host Specificity of Campylobacter jejuni. Sci. Rep. 2021, 11, 10358. [Google Scholar] [CrossRef] [PubMed]
- Bolton, D.J. Campylobacter Virulence and Survival Factors. Food Microbiol. 2015, 48, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Kirk, M.D.; Pires, S.M.; Black, R.E.; Caipo, M.; Crump, J.A.; Devleesschauwer, B.; Döpfer, D.; Fazil, A.; Fischer-Walker, C.L.; Hald, T.; et al. World Health Organization Estimates of the Global and Regional Disease Burden of 22 Foodborne Bacterial, Protozoal, and Viral Diseases, 2010: A Data Synthesis. PLoS Med. 2015, 12, e1001921. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, H.L.; Dalager-Pedersen, M.; Nielsen, H. Risk of Inflammatory Bowel Disease after Campylobacter jejuni and Campylobacter concisus Infection: A Population-Based Cohort Study. Scand. J. Gastroenterol. 2019, 54, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, Y.; Rojas, M.; Pacheco, Y.; Acosta-Ampudia, Y.; Ramírez-Santana, C.; Monsalve, D.M.; Gershwin, M.E.; Anaya, J.-M. Guillain–Barré Syndrome, Transverse Myelitis and Infectious Diseases. Cell. Mol. Immunol. 2018, 15, 547–562. [Google Scholar] [CrossRef] [Green Version]
- Leonhard, S.E.; Mandarakas, M.R.; Gondim, F.A.A.; Bateman, K.; Ferreira, M.L.B.; Cornblath, D.R.; van Doorn, P.A.; Dourado, M.E.; Hughes, R.A.C.; Islam, B.; et al. Diagnosis and Management of Guillain–Barré Syndrome in Ten Steps. Nat. Rev. Neurol. 2019, 15, 671–683. [Google Scholar] [CrossRef]
- Gaytán, M.O.; Martínez-Santos, V.I.; Soto, E.; González-Pedrajo, B. Type Three Secretion System in Attaching and Effacing Pathogens. Front. Cell. Infect. Microbiol. 2016, 6, 129. [Google Scholar] [CrossRef] [Green Version]
- Burnham, P.M.; Hendrixson, D.R. Campylobacter jejuni: Collective Components Promoting a Successful Enteric Lifestyle. Nat. Rev. Microbiol. 2018, 16, 551–565. [Google Scholar] [CrossRef]
- Dos Santos, A.M.P.; Ferrari, R.G.; Conte-Junior, C.A. Virulence Factors in Salmonella Typhimurium: The Sagacity of a Bacterium. Curr. Microbiol. 2019, 76, 762–773. [Google Scholar] [CrossRef]
- Elmi, A.; Nasher, F.; Dorrell, N.; Wren, B.; Gundogdu, O. Revisiting Campylobacter jejuni Virulence and Fitness Factors: Role in Sensing, Adapting, and Competing. Front. Cell. Infect. Microbiol. 2021, 10, 607704. [Google Scholar] [CrossRef] [PubMed]
- Mehat, J.W.; Park, S.F.; van Vliet, A.H.M.; La Ragione, R.M. CapC, a Novel Autotransporter and Virulence Factor of Campylobacter jejuni. Appl. Environ. Microbiol. 2018, 84, e01032-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehat, J.W.; La Ragione, R.M.; van Vliet, A.H.M. Campylobacter jejuni and Campylobacter coli Autotransporter Genes Exhibit Lineage-Associated Distribution and Decay. BMC Genom. 2020, 21, 314. [Google Scholar] [CrossRef] [PubMed]
- Gripp, E.; Hlahla, D.; Didelot, X.; Kops, F.; Maurischat, S.; Tedin, K.; Alter, T.; Ellerbroek, L.; Schreiber, K.; Schomburg, D.; et al. Closely Related Campylobacter jejuni Strains from Different Sources Reveal a Generalist Rather than a Specialist Lifestyle. BMC Genom. 2011, 12, 584. [Google Scholar] [CrossRef] [Green Version]
- Colles, F.M.; Maiden, M.C.J. Campylobacter Sequence Typing Databases: Applications and Future Prospects. Microbiology 2012, 158, 2695–2709. [Google Scholar] [CrossRef] [PubMed]
- Wallace, R.L.; Cribb, D.M.; Bulach, D.M.; Ingle, D.J.; Joensen, K.G.; Nielsen, E.M.; Leekitcharoenphon, P.; Stingl, K.; Kirk, M.D. Campylobacter jejuni ST50, a Pathogen of Global Importance: A Comparative Genomic Analysis of Isolates from Australia, Europe and North America. Zoonoses Public Health 2021, 68, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2017, 45, D37–D42. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, A.M.P.; Ferrari, R.G.; Panzenhagen, P.; Rodrigues, G.L.; Conte-Junior, C.A. Virulence Genes Identification and Characterization Revealed the Presence of the Yersinia High Pathogenicity Island (HPI) in Salmonella from Brazil. Gene 2021, 787, 145646. [Google Scholar] [CrossRef]
- De Fátima RauberWürfel, S.; Jorge, S.; de Oliveira, N.R.; Kremer, F.S.; Sanchez, C.D.; Campos, V.F.; da Silva Pinto, L.; da Silva, W.P.; Dellagostin, O.A. Campylobacter jejuni Isolated from Poultry Meat in Brazil: In Silico Analysis and Genomic Features of Two Strains with Different Phenotypes of Antimicrobial Susceptibility. Mol. Biol. Rep. 2020, 47, 671–681. [Google Scholar] [CrossRef]
- MalbergTetzschner, A.M.; Johnson, J.R.; Johnston, B.D.; Lund, O.; Scheutz, F. In Silico Genotyping of Escherichia coli Isolates for Extraintestinal Virulence Genes by Use of Whole-Genome Sequencing Data. J. Clin. Microbiol. 2020, 58, e01269-20. [Google Scholar] [CrossRef]
- Rodrigues, G.L.; Panzenhagen, P.; Ferrari, R.G.; dos Santos, A.; Paschoalin, V.M.F.; Conte-Junior, C.A. Frequency of Antimicrobial Resistance Genes in Salmonella From Brazil by in Silico Whole-Genome Sequencing Analysis: An Overview of the Last Four Decades. Front. Microbiol. 2020, 11, 1864. [Google Scholar] [CrossRef]
- Chen, L. VFDB: A Reference Database for Bacterial Virulence Factors. Nucleic Acids Res. 2004, 33, D325–D328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and Refined Dataset for Big Data Analysis—10 Years On. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef] [PubMed]
- Hasman, H.; Saputra, D.; Sicheritz-Ponten, T.; Lund, O.; Svendsen, C.A.; Frimodt-Møller, N.; Aarestrup, F.M. Rapid Whole-Genome Sequencing for Detection and Characterization of Microorganisms Directly from Clinical Samples. J. Clin. Microbiol. 2014, 52, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, M.V.; Cosentino, S.; Lukjancenko, O.; Saputra, D.; Rasmussen, S.; Hasman, H.; Sicheritz-Pontén, T.; Aarestrup, F.M.; Ussery, D.W.; Lund, O. Benchmarking of Methods for Genomic Taxonomy. J. Clin. Microbiol. 2014, 52, 1529–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clausen, P.T.L.C.; Aarestrup, F.M.; Lund, O. Rapid and Precise Alignment of Raw Reads against Redundant Databases with KMA. BMC Bioinform. 2018, 19, 307. [Google Scholar] [CrossRef] [PubMed]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-Access Bacterial Population Genomics: BIGSdb Software, the PubMLST.Org Website and Their Applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef] [PubMed]
- Clausen, P.T.L.C.; Zankari, E.; Aarestrup, F.M.; Lund, O. Benchmarking of Methods for Identification of Antimicrobial Resistance Genes in Bacterial Whole Genome Data. J. Antimicrob. Chemother. 2016, 71, 2484–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meinersmann, R.J.; Helsel, L.O.; Fields, P.I.; Hiett, K.L. Discrimination of Campylobacter jejuni Isolates by Fla Gene Sequencing. J. Clin. Microbiol. 1997, 35, 2810–2814. [Google Scholar] [CrossRef] [Green Version]
- Cody, A.J.; Maiden, M.J.C.; Dingle, K.E. Genetic Diversity and Stability of the PorA Allele as a Genetic Marker in Human Campylobacter Infection. Microbiology 2009, 155, 4145–4154. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Skirrow, M.B. Campylobacter Enteritis: A “New” Disease. Br. Med. J. 1977, 2, 9–11. [Google Scholar] [CrossRef] [Green Version]
- Skirrow, M.B. John McFadyean and the Centenary of the First Isolation of Campylobacter Species. Clin. Infect. Dis. 2006, 43, 1213–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mourkas, E.; Taylor, A.J.; Méric, G.; Bayliss, S.C.; Pascoe, B.; Mageiros, L.; Calland, J.K.; Hitchings, M.D.; Ridley, A.; Vidal, A.; et al. Agricultural Intensification and the Evolution of Host Specialism in the Enteric Pathogen Campylobacter jejuni. Proc. Natl. Acad. Sci. USA 2020, 117, 11018–11028. [Google Scholar] [CrossRef]
- Maiden, M.C.J.; van Rensburg, M.J.J.; Bray, J.E.; Earle, S.G.; Ford, S.A.; Jolley, K.A.; McCarthy, N.D. MLST Revisited: The Gene-by-Gene Approach to Bacterial Genomics. Nat. Rev. Microbiol. 2013, 11, 728–736. [Google Scholar] [CrossRef] [Green Version]
- Dingle, K.E.; Colles, F.M.; Wareing, D.R.A.; Ure, R.; Fox, A.J.; Bolton, F.E.; Bootsma, H.J.; Willems, R.J.L.; Urwin, R.; Maiden, M.C.J. Multilocus Sequence Typing System for Campylobacter jejuni. J. Clin. Microbiol. 2001, 39, 14–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheppard, S.K.; Cheng, L.; Méric, G.; Haan, C.P.A.; Llarena, A.; Marttinen, P.; Vidal, A.; Ridley, A.; Clifton-Hadley, F.; Connor, T.R.; et al. Cryptic Ecology among Host Generalist Campylobacter jejuni in Domestic Animals. Mol. Ecol. 2014, 23, 2442–2451. [Google Scholar] [CrossRef] [Green Version]
- Sheppard, S.K.; Colles, F.; Richardson, J.; Cody, A.J.; Elson, R.; Lawson, A.; Brick, G.; Meldrum, R.; Little, C.L.; Owen, R.J.; et al. Host Association of Campylobacter Genotypes Transcends Geographic Variation. Appl. Environ. Microbiol. 2010, 76, 5269–5277. [Google Scholar] [CrossRef] [Green Version]
- Dearlove, B.L. Rapid Host Switching in Generalist Campylobacter Strains Erodes the Signal for Tracing Human Infections. ISME J. 2016, 10, 721–729. [Google Scholar] [CrossRef] [Green Version]
- Mouftah, S.F.; Cobo-Díaz, J.F.; Álvarez-Ordóñez, A.; Mousa, A.; Calland, J.K.; Pascoe, B.; Sheppard, S.K.; Elhadidy, M. Stress Resistance Associated with Multi-Host Transmission and Enhanced Biofilm Formation at 42 °C among Hyper-Aerotolerant Generalist Campylobacter jejuni. Food Microbiol. 2021, 95, 103706. [Google Scholar] [CrossRef] [PubMed]
- Karlyshev, A.V.; Champion, O.L.; Churcher, C.; Brisson, J.-R.; Jarrell, H.C.; Gilbert, M.; Brochu, D.; St Michael, F.; Li, J.; Wakarchuk, W.W.; et al. Analysis of Campylobacter jejuni Capsular Loci Reveals Multiple Mechanisms for the Generation of Structural Diversity and the Ability to Form Complex Heptoses: C. jejuni CPS Biosynthesis. Mol. Microbiol. 2004, 55, 90–103. [Google Scholar] [CrossRef] [PubMed]
- McNally, D.J.; Hui, J.P.M.; Aubry, A.J.; Mui, K.K.K.; Guerry, P.; Brisson, J.-R.; Logan, S.M.; Soo, E.C. Functional Characterization of the Flagellar Glycosylation Locus in Campylobacter jejuni 81–176 Using a Focused Metabolomics Approach. J. Biol. Chem. 2006, 281, 18489–18498. [Google Scholar] [CrossRef] [Green Version]
- Dzieciatkowska, M.; Brochu, D.; van Belkum, A.; Heikema, A.P.; Yuki, N.; Houliston, R.S.; Richards, J.C.; Gilbert, M.; Li, J. Mass Spectrometric Analysis of Intact Lipooligosaccharide: Direct Evidence for O-Acetylated Sialic Acids and Discovery of O-Linked Glycine Expressed by Campylobacter jejuni. Biochemistry 2007, 46, 14704–14714. [Google Scholar] [CrossRef]
- Karlyshev, A.V.; Linton, D.; Gregson, N.A.; Lastovica, A.J.; Wren, B.W. Genetic and Biochemical Evidence of a Campylobacter jejuni Capsular Polysaccharide That Accounts for Penner Serotype Specificity: Genetics and Biochemistry of C. jejuni LPS Biosynthesis. Mol. Microbiol. 2002, 35, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Bacon, D.J.; Szymanski, C.M.; Burr, D.H.; Silver, R.P.; Alm, R.A.; Guerry, P. A Phase-Variable Capsule Is Involved in Virulence of Campylobacter jejuni 81-176: C. jejuni Capsular Phase Variation. Mol. Microbiol. 2001, 40, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Sahin, O.; Terhorst, S.A.; Burrough, E.R.; Shen, Z.; Wu, Z.; Dai, L.; Tang, Y.; Plummer, P.J.; Ji, J.; Yaeger, M.J.; et al. Key Role of Capsular Polysaccharide in the Induction of Systemic Infection and Abortion by Hypervirulent Campylobacter jejuni. Infect. Immun. 2017, 85, e00001-17. [Google Scholar] [CrossRef] [Green Version]
- Stahl, M.; Ries, J.; Vermeulen, J.; Yang, H.; Sham, H.P.; Crowley, S.M.; Badayeva, Y.; Turvey, S.E.; Gaynor, E.C.; Li, X.; et al. A Novel Mouse Model of Campylobacter jejuni Gastroenteritis Reveals Key Pro-Inflammatory and Tissue Protective Roles for Toll-like Receptor Signaling during Infection. PLoS Pathog. 2014, 10, e1004264. [Google Scholar] [CrossRef] [Green Version]
- Christie, P.J. Type IV Secretion: Intercellular Transfer of Macromolecules by Systems Ancestrally Related to Conjugation Machines. Mol. Microbiol. 2001, 40, 294–305. [Google Scholar] [CrossRef] [Green Version]
- Tracz, D.M.; Keelan, M.; Ahmed-Bentley, J.; Gibreel, A.; Kowalewska-Grochowska, K.; Taylor, D.E. PVir and Bloody Diarrhea in Campylobacter jejuni Enteritis. Emerg. Infect. Dis. 2005, 11, 839–843. [Google Scholar] [CrossRef]
- Bacon, D.J.; Alm, R.A.; Burr, D.H.; Hu, L.; Kopecko, D.J.; Ewing, C.P.; Trust, T.J.; Guerry, P. Involvement of a Plasmid in Virulence of Campylobacter jejuni 81-176. Infect. Immun. 2000, 68, 4384–4390. [Google Scholar] [CrossRef] [Green Version]
- Louwen, R.P.L.; van Belkum, A.; Wagenaar, J.A.; Doorduyn, Y.; Achterberg, R.; Endtz, H.P. Lack of Association between the Presence of the PVir Plasmid and Bloody Diarrhea in Campylobacter jejuni Enteritis. J. Clin. Microbiol. 2006, 44, 1867–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marasini, D.; Fakhr, M. Exploring PFGE for Detecting Large Plasmids in Campylobacter jejuni and Campylobacter coli Isolated from Various Retail Meats. Pathogens 2014, 3, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iglesias-Torrens, Y.; Miró, E.; Guirado, P.; Llovet, T.; Muñoz, C.; Cerdà-Cuéllar, M.; Madrid, C.; Balsalobre, C.; Navarro, F. Population Structure, Antimicrobial Resistance, and Virulence-Associated Genes in Campylobacter jejuni Isolated from Three Ecological Niches: Gastroenteritis Patients, Broilers, and Wild Birds. Front. Microbiol. 2018, 9, 1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt-Ott, R.; Pohl, S.; Burghard, S.; Weig, M.; Groß, U. Identification and Characterization of a Major Subgroup of Conjugative Campylobacter jejuni Plasmids. J. Infect. 2005, 50, 12–21. [Google Scholar] [CrossRef]
- Nachamkin, I.; Bohachick, K.; Patton, C.M. Flagellin Gene Typing of Campylobacter jejuni by Restriction Fragment Length Polymorphism Analysis. J. Clin. Microbiol. 1993, 31, 1531–1536. [Google Scholar] [CrossRef] [Green Version]
- Harrington, C.S.; Thomson-Carter, F.M.; Carter, P.E. Evidence for Recombination in the Flagellin Locus of Campylobacter jejuni: Implications for the Flagellin Gene Typing Scheme. J. Clin. Microbiol. 1997, 35, 2386–2392. [Google Scholar] [CrossRef] [Green Version]
- Wassenaar, T.M.; Bleumink-Pluym, N.M.; van der Zeijst, B.A. Inactivation of Campylobacter jejuni Flagellin Genes by Homologous Recombination Demonstrates That flaA but Not flaB Is Required for Invasion. EMBO J. 1991, 10, 2055–2061. [Google Scholar] [CrossRef]
- Wirz, S.E.; Overesch, G.; Kuhnert, P.; Korczak, B.M. Genotype and Antibiotic Resistance Analyses of Campylobacter Isolates from Ceca and Carcasses of Slaughtered Broiler Flocks. Appl. Environ. Microbiol. 2010, 76, 6377–6386. [Google Scholar] [CrossRef] [Green Version]
- Magnússon, S.H.; Guðmundsdóttir, S.; Reynisson, E.; Rúnarsson, Á.R.; Harðardóttir, H.; Gunnarson, E.; Georgsson, F.; Reiersen, J.; Marteinsson, V.T. Comparison of Campylobacter jejuni Isolates from Human, Food, Veterinary and Environmental Sources in Iceland Using PFGE, MLST and Fla-SVR Sequencing. J. Appl. Microbiol. 2011, 111, 971–981. [Google Scholar] [CrossRef]
- Gomes, C.N.; Souza, R.A.; Passaglia, J.; Duque, S.S.; Medeiros, M.I.C.; Falcão, J.P. Genotyping of Campylobacter coli Strains Isolated in Brazil Suggests Possible Contamination amongst Environmental, Human, Animal and Food Sources. J. Med. Microbiol. 2016, 65, 80–90. [Google Scholar] [CrossRef]
- Talukder, K.A.; Aslam, M.; Islam, Z.; Azmi, I.J.; Dutta, D.K.; Hossain, S.; Nur-E-Kamal, A.; Nair, G.B.; Cravioto, A.; Sack, D.A.; et al. Prevalence of Virulence Genes and Cytolethal Distending Toxin Production in Campylobacter jejuni Isolates from Diarrheal Patients in Bangladesh. J. Clin. Microbiol. 2008, 46, 1485–1488. [Google Scholar] [CrossRef] [Green Version]
- Wieczorek, K.; Szewczyk, R.; Osek, J. Prevalence, Antimicrobial Resistance, and Molecular Characterization of Campylobacter jejuni and C. coli Isolated from Retail Raw Meat in Poland. Vet. Med. 2012, 57, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Casabonne, C.; Gonzalez, A.; Aquili, V.; Subils, T.; Balague, C. Prevalence of Seven Virulence Genes of Campylobacter jejuni Isolated from Patients with Diarrhea in Rosario, Argentina. Int. J. Infect. 2016, 3, e37727. [Google Scholar] [CrossRef]
- Hanning, I.; Biswas, D.; Herrera, P.; Roesler, M.; Ricke, S.C. Prevalence and Characterization of Campylobacter jejuni Isolated from Pasture Flock Poultry. J. Food Sci. 2010, 75, M496–M502. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Jang, S.S.; Choo, E.; Heu, S.; Ryu, S. Prevalence, Genetic Diversity, and Antibiotic Resistance Patterns of Campylobacter jejuni from Retail Raw Chickens in Korea. Int. J. Food Microbiol. 2007, 114, 50–59. [Google Scholar] [CrossRef]
- Melo, R.T.; Grazziotin, A.L.; Júnior, E.C.V.; Prado, R.R.; Mendonça, E.P.; Monteiro, G.P.; Peres, P.A.B.M.; Rossi, D.A. Evolution of Campylobacter jejuni of Poultry Origin in Brazil. Food Microbiol. 2019, 82, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Rossler, E.; Olivero, C.; Soto, L.P.; Frizzo, L.S.; Zimmermann, J.; Rosmini, M.R.; Sequeira, G.J.; Signorini, M.L.; Zbrun, M.V. Prevalence, Genotypic Diversity and Detection of Virulence Genes in Thermotolerant Campylobacter at Different Stages of the Poultry Meat Supply Chain. Int. J. Food Microbiol. 2020, 326, 108641. [Google Scholar] [CrossRef]
- Abd El-Hamid, M.I.; Abd El-Aziz, N.K.; Samir, M.; El-Naenaeey, E.Y.; Abo Remela, E.M.; Mosbah, R.A.; Bendary, M.M. Genetic Diversity of Campylobacter jejuni Isolated From Avian and Human Sources in Egypt. Front. Microbiol. 2019, 10, 2353. [Google Scholar] [CrossRef]
- Ammar, A.M.; El-Naenaeey, E.-S.Y.; El-Malt, R.M.S.; El-Gedawy, A.A.; Khalifa, E.; Elnahriry, S.S.; Abd El-Hamid, M.I. Prevalence, Antimicrobial Susceptibility, Virulence and Genotyping of Campylobacter jejuni with a Special Reference to the Anti-Virulence Potential of Eugenol and β-Resorcylic Acid on Some Multi-Drug Resistant Isolates in Egypt. Animals 2020, 11, 3. [Google Scholar] [CrossRef]
- Han, X.; Guan, X.; Zeng, H.; Li, J.; Huang, X.; Wen, Y.; Zhao, Q.; Huang, X.; Yan, Q.; Huang, Y.; et al. Prevalence, Antimicrobial Resistance Profiles and Virulence-Associated Genes of Thermophilic Campylobacter spp. Isolated from Ducks in a Chinese Slaughterhouse. Food Control 2019, 104, 157–166. [Google Scholar] [CrossRef]
- Bravo, V.; Katz, A.; Porte, L.; Weitzel, T.; Varela, C.; Gonzalez-Escalona, N.; Blondel, C.J. Genomic Analysis of the Diversity, Antimicrobial Resistance and Virulence Potential of Clinical Campylobacter jejuni and Campylobacter coli Strains from Chile. PLoS Negl. Trop. Dis. 2021, 15, e0009207. [Google Scholar] [CrossRef]
- Truccollo, B.; Whyte, P.; Burgess, C.; Bolton, D. Genetic Characterisation of a Subset of Campylobacter jejuni Isolates from Clinical and Poultry Sources in Ireland. PLoS ONE 2021, 16, e0246843. [Google Scholar] [CrossRef]
- Fiedoruk, K.; Daniluk, T.; Rozkiewicz, D.; Oldak, E.; Prasad, S.; Swiecicka, I. Whole-Genome Comparative Analysis of Campylobacter jejuni Strains Isolated from Patients with Diarrhea in Northeastern Poland. Gut Pathog. 2019, 11, 32. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Xu, H.; Ning, C.; Xiang, L.; Ren, Q.; Zhang, T.; Zhang, Y.; Gao, R. Multi-Omics Approach Reveals the Potential Core Vaccine Targets for the Emerging Foodborne Pathogen Campylobacter jejuni. Front. Microbiol. 2021, 12, 665858. [Google Scholar] [CrossRef]
- Alm, R.A.; Guerry, P.; Trust, T.J. Significance of Duplicated Flagellin Genes in Campylobacter. J. Mol. Biol. 1993, 230, 359–363. [Google Scholar] [CrossRef] [Green Version]
- Santoyo, G.; Romero, D. Gene Conversion and Concerted Evolution in Bacterial Genomes. FEMS Microbiol. Rev. 2005, 29, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Meinersmann, R.J.; Hiett, K.L. Concerted Evolution of Duplicate Fla Genes in Campylobacter. Microbiology 2000, 146, 2283–2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magana, M.; Chatzipanagiotou, S.; Burriel, A.R.; Ioannidis, A. Inquiring into the Gaps of Campylobacter Surveillance Methods. Vet. Sci. 2017, 4, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linton, D.; Gilbert, M.; Hitchen, P.G.; Dell, A.; Morris, H.R.; Wakarchuk, W.W.; Gregson, N.A.; Wren, B.W. Phase Variation of a β-1,3 Galactosyltransferase Involved in Generation of the Ganglioside GM1-like Lipo-Oligosaccharide of Campylobacter jejuni: C. jejuni LOS Phase Variation. Mol. Microbiol. 2002, 37, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Koga, M.; Gilbert, M.; Takahashi, M.; Li, J.; Koike, S.; Hirata, K.; Yuki, N. Comprehensive Analysis of Bacterial Risk Factors for the Development of Guillain-Barré Syndrome after Campylobacter jejuni Enteritis. J. Infect. Dis. 2006, 193, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Guirado, P.; Paytubi, S.; Miró, E.; Iglesias-Torrens, Y.; Navarro, F.; Cerdà-Cuéllar, M.; Stephan-Otto Attolini, C.; Balsalobre, C.; Madrid, C. Differential Distribution of the WlaN and CgtB Genes, Associated with Guillain-Barré Syndrome, in Campylobacter jejuni Isolates from Humans, Broiler Chickens, and Wild Birds. Microorganisms 2020, 8, 325. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.; Niwa, H.; Itoh, K. Prevalence of 11 Pathogenic Genes of Campylobacter jejuni by PCR in Strains Isolated from Humans, Poultry Meat and Broiler and Bovine Faeces. J. Med. Microbiol. 2003, 52, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Koolman, L.; Whyte, P.; Burgess, C.; Bolton, D. Distribution of Virulence-Associated Genes in a Selection of Campylobacter Isolates. Foodborne Pathog. Dis. 2015, 12, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Sierra-Arguello, Y.M.; Perdoncini, G.; Rodrigues, L.B.; Ruschel dos Santos, L.; Apellanis Borges, K.; QuediFurian, T.; Pippi Salle, C.T.; de Souza Moraes, H.L.; Pereira Gomes, M.J.; Pinheiro do Nascimento, V. Identification of Pathogenic Genes in Campylobacter jejuni Isolated from Broiler Carcasses and Broiler Slaughterhouses. Sci. Rep. 2021, 11, 4588. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Lee, M.Y.; Kim, S.J.; Jeon, S.-E.; Cha, I.; Hong, S.; Chung, G.T.; Huh, M.-J.; Kang, Y.-H.; Yoo, C.-K.; et al. High-Level Ciprofloxacin-Resistant Campylobacter jejuni Isolates Circulating in Humans and Animals in Incheon, Republic of Korea. Zoonoses Public Health 2016, 63, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-S.; Park, C.; Kim, Y.-J. Role of FlgA for Flagellar Biosynthesis and Biofilm Formation of Campylobacter jejuni NCTC11168. J. Microbiol. Biotechnol. 2015, 25, 1871–1879. [Google Scholar] [CrossRef]
- Neal-McKinney, J.M.; Konkel, M.E. The Campylobacter jejuni CiaC Virulence Protein Is Secreted from the Flagellum and Delivered to the Cytosol of Host Cells. Front. Cell. Infect. Microbiol. 2012, 2, 31. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Rank | North America (n = 30,504) | Europe (n = 9246) | South America (n = 191) | Oceania (n = 171) | Asia (n = 168) | Africa (n = 91) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ST | n (%) | ST | n (%) | ST | n (%) | ST | n (%) | ST | n (%) | ST | n (%) | |
| 1 | 353 | 1602 (5.25) | 50 | 563 (6.09) | 353 | 24 (12.57) | 50 | 29 (16.96) | 2988 | 14 (8.33) | 353 | 9 (9.78) |
| 2 | 50 | 1388 (4.55) | 5136 | 416 (4.50) | 2993 | 20 (10.47) | 528 | 13 (7.60) | 22 | 8 (4.76) | 362 | 5 (5.43) |
| 3 | 48 | 1260 (4.10) | 48 | 398 (4.30) | 8741 | 15 (7.85) | 48 | 11 (6.43) | 985 | 8 (4.76) | 7784 | 5 (5.43) |
| 4 | 45 | 1236 (4.05) | 21 | 383 (4.14) | 1359 | 14 (7.33) | 567 | 7 (4.09) | 587 | 7 (4.17) | 52 | 4 (4.35) |
| 5 | 982 | 1220 (3.99) | 45 | 297 (3.21) | 475 | 12 (6.28) | 9817 | 6 (3.51) | 2140 | 6 (3.57) | 460 | 4 (4.35) |
| 6 | 8 | 1013 (3.32) | 257 | 273 (2.95) | 50 | 7 (3.66) | 2398 | 6 (3.51) | 10,086 | 5 (2.98) | 1038 | 4 (4.35) |
| 7 | 806 | 857 (2.80) | 122 | 239 (2.58) | 52 | 7 (3.66) | 658 | 5 (2.92) | 27 | 4 (2.38) | 5 | 3 (3.26) |
| 8 | 459 | 756 (2.47) | 19 | 237 (2.56) | 403 | 6 (3.14) | 257 | 5 (2.92) | 986 | 4 (2.38) | 523 | 3 (3.26) |
| 9 | 21 | 697 (2.28) | 61 | 209 (2.26) | 463 | 6 (3.14) | 190 | 5 (2.92) | 2209 | 4 (2.38) | 607 | 3 (3.26) |
| 10 | 222 | 674 (2.20) | 354 | 171 (1.85) | 883 | 4 (2.09) | 6964 | 4 (2.34) | 5 | 3 (1.79) | 1036 | 3 (3.26) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panzenhagen, P.; Portes, A.B.; dos Santos, A.M.P.; Duque, S.d.S.; Conte Junior, C.A. The Distribution of Campylobacter jejuni Virulence Genes in Genomes Worldwide Derived from the NCBI Pathogen Detection Database. Genes 2021, 12, 1538. https://doi.org/10.3390/genes12101538
Panzenhagen P, Portes AB, dos Santos AMP, Duque SdS, Conte Junior CA. The Distribution of Campylobacter jejuni Virulence Genes in Genomes Worldwide Derived from the NCBI Pathogen Detection Database. Genes. 2021; 12(10):1538. https://doi.org/10.3390/genes12101538
Chicago/Turabian StylePanzenhagen, Pedro, Ana Beatriz Portes, Anamaria M. P. dos Santos, Sheila da Silva Duque, and Carlos Adam Conte Junior. 2021. "The Distribution of Campylobacter jejuni Virulence Genes in Genomes Worldwide Derived from the NCBI Pathogen Detection Database" Genes 12, no. 10: 1538. https://doi.org/10.3390/genes12101538
APA StylePanzenhagen, P., Portes, A. B., dos Santos, A. M. P., Duque, S. d. S., & Conte Junior, C. A. (2021). The Distribution of Campylobacter jejuni Virulence Genes in Genomes Worldwide Derived from the NCBI Pathogen Detection Database. Genes, 12(10), 1538. https://doi.org/10.3390/genes12101538