
Inherited Proteoglycan Biosynthesis Defects—Current Laboratory Tools and Bikunin as a Promising Blood Biomarker

 and
and 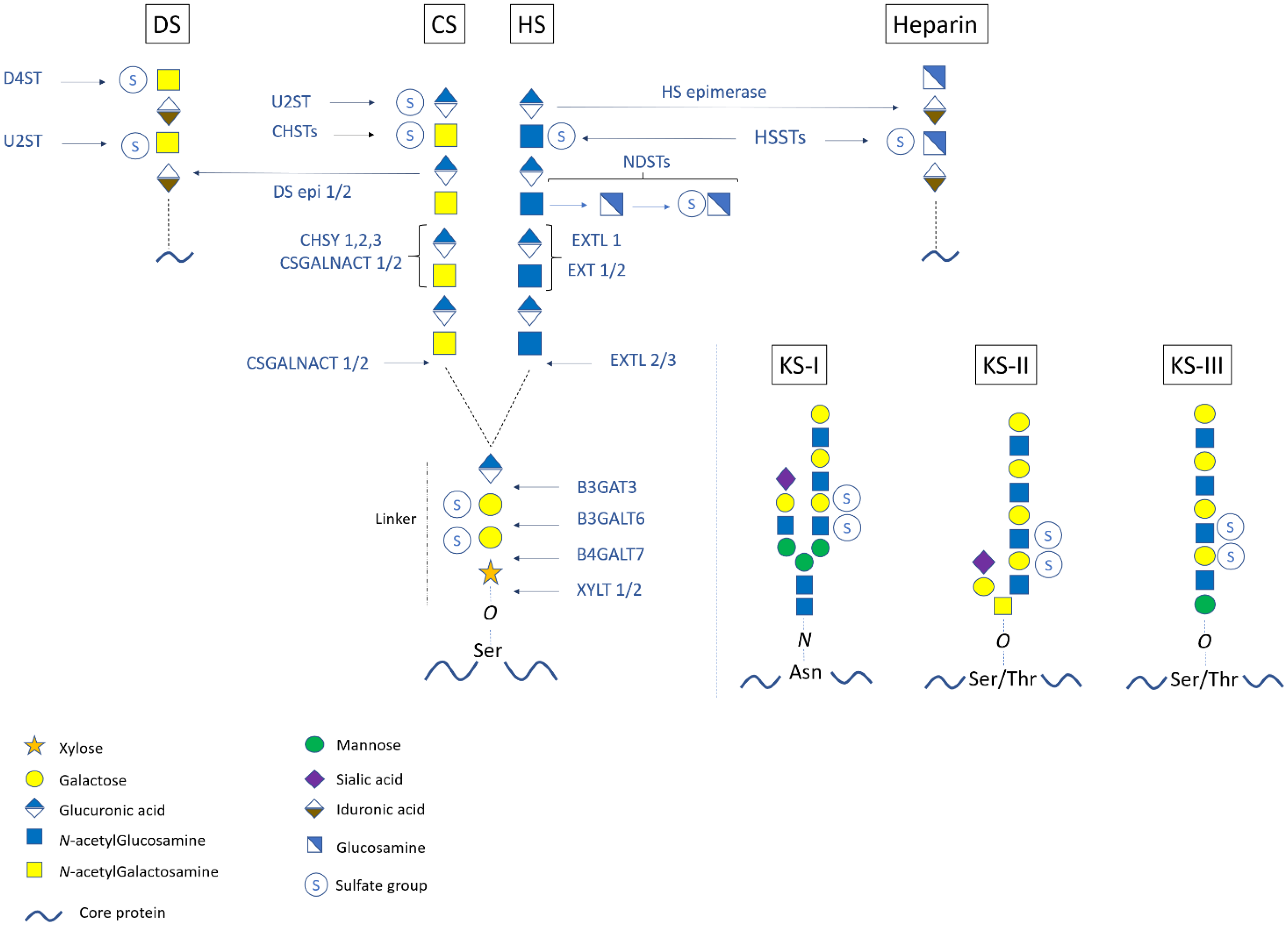{kind=link}
{kind=link}
{kind=link}
{kind=link}
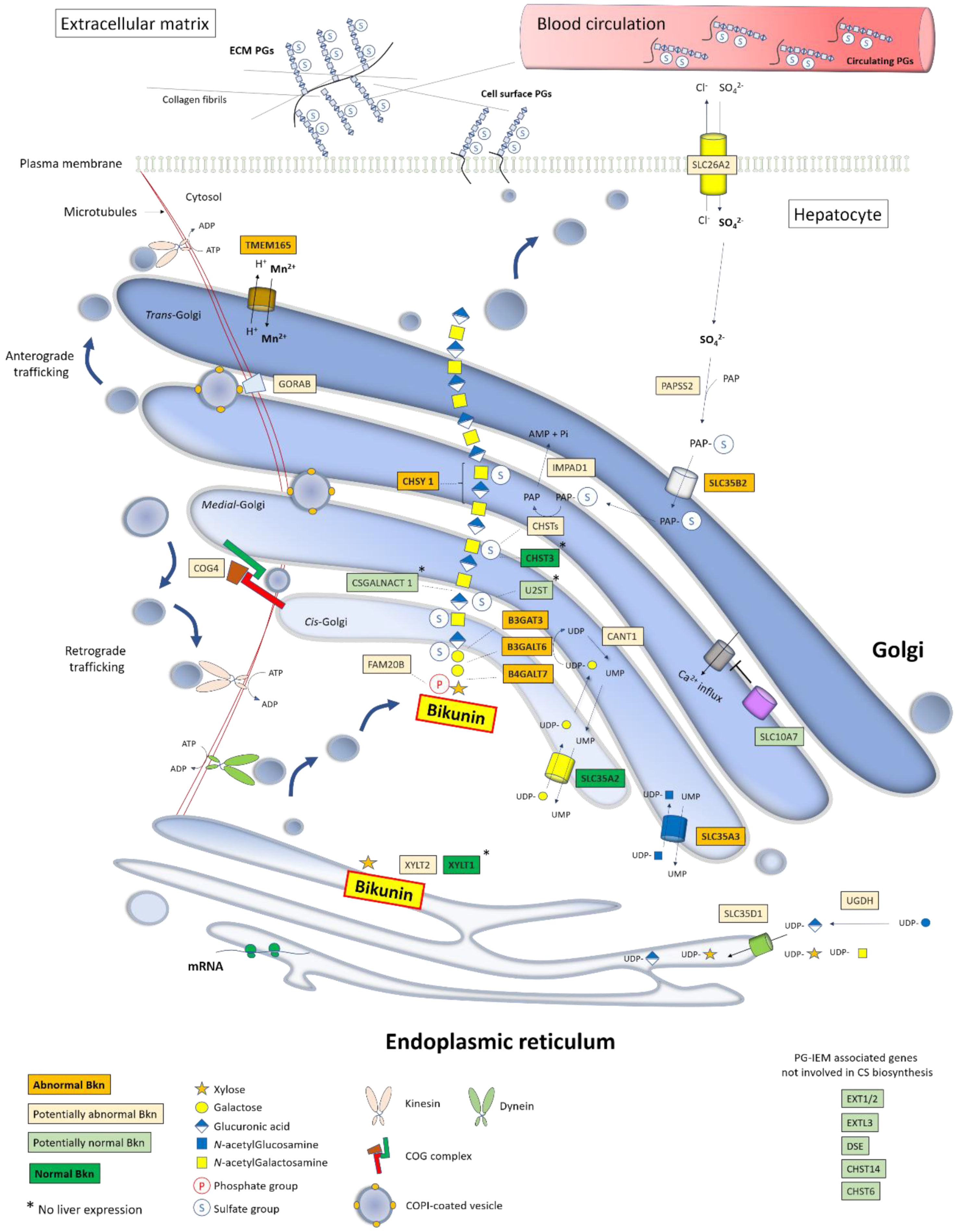{kind=link}
Abstract
:1. Introduction
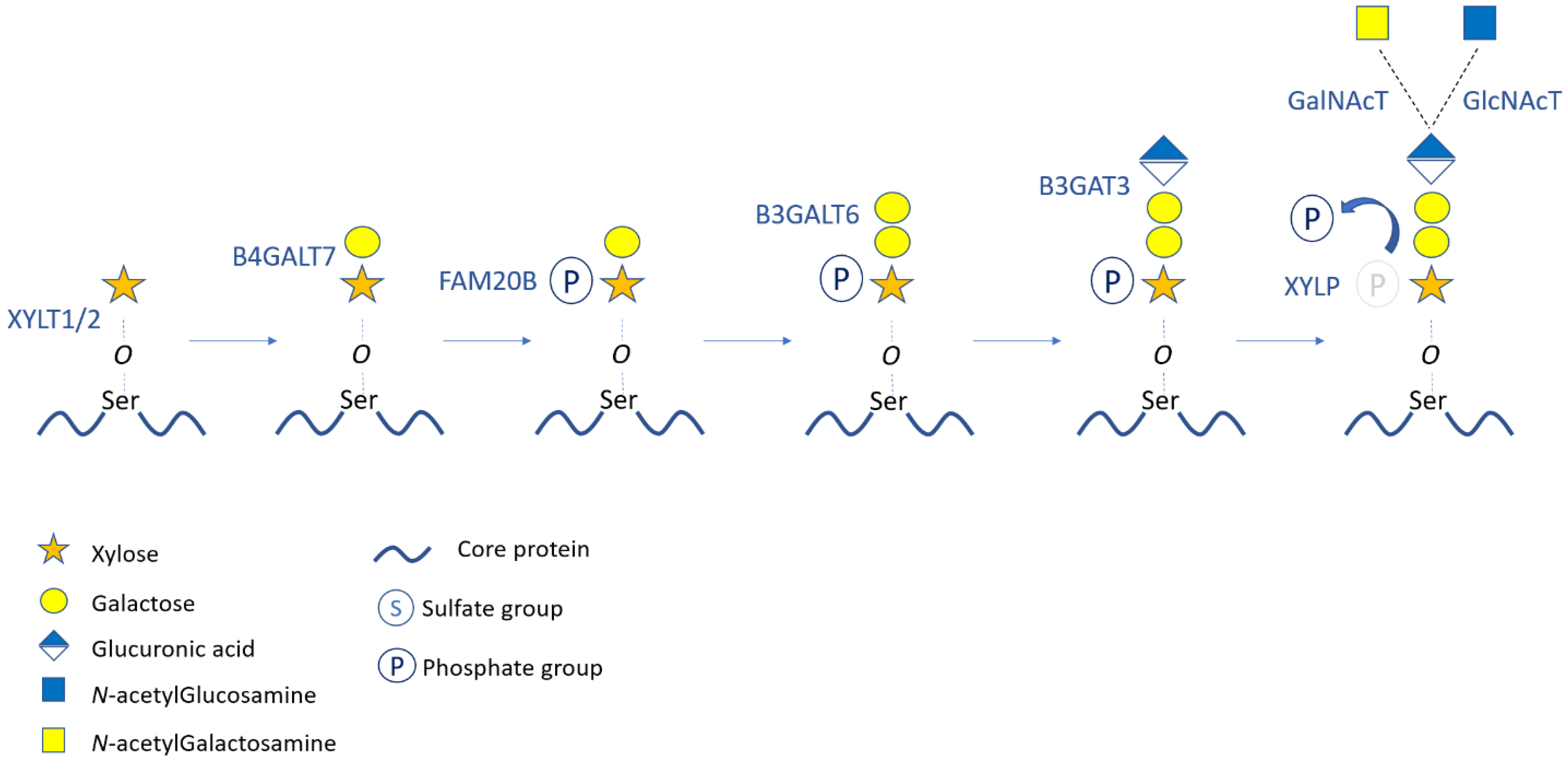
2. Structure, Synthesis, and Modifications of Proteoglycans
3. Classification, Distribution, and Roles of Proteoglycans
4. Inborn Errors of Proteoglycan Metabolism
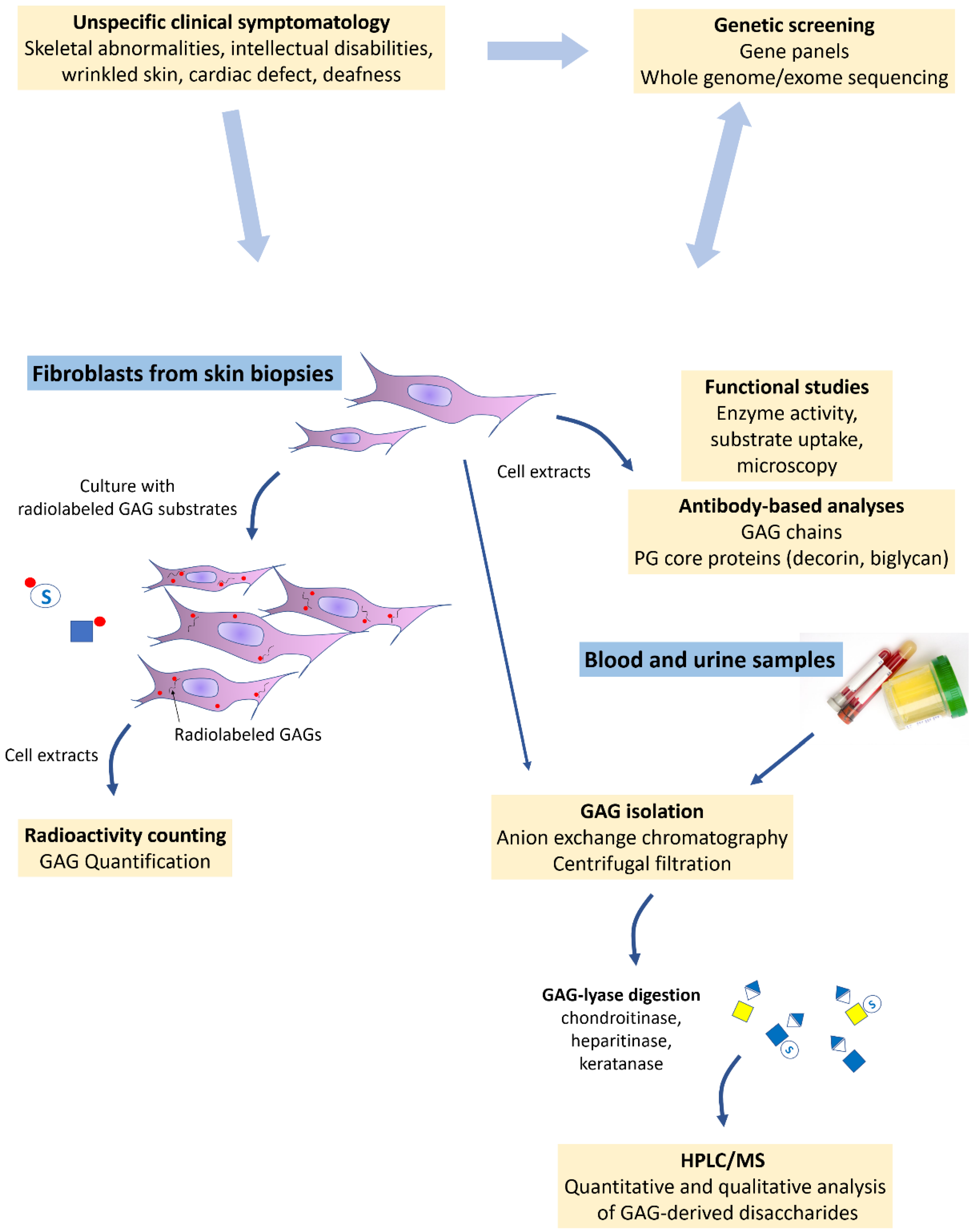
5. Current Laboratory Tools and Examples of Applications
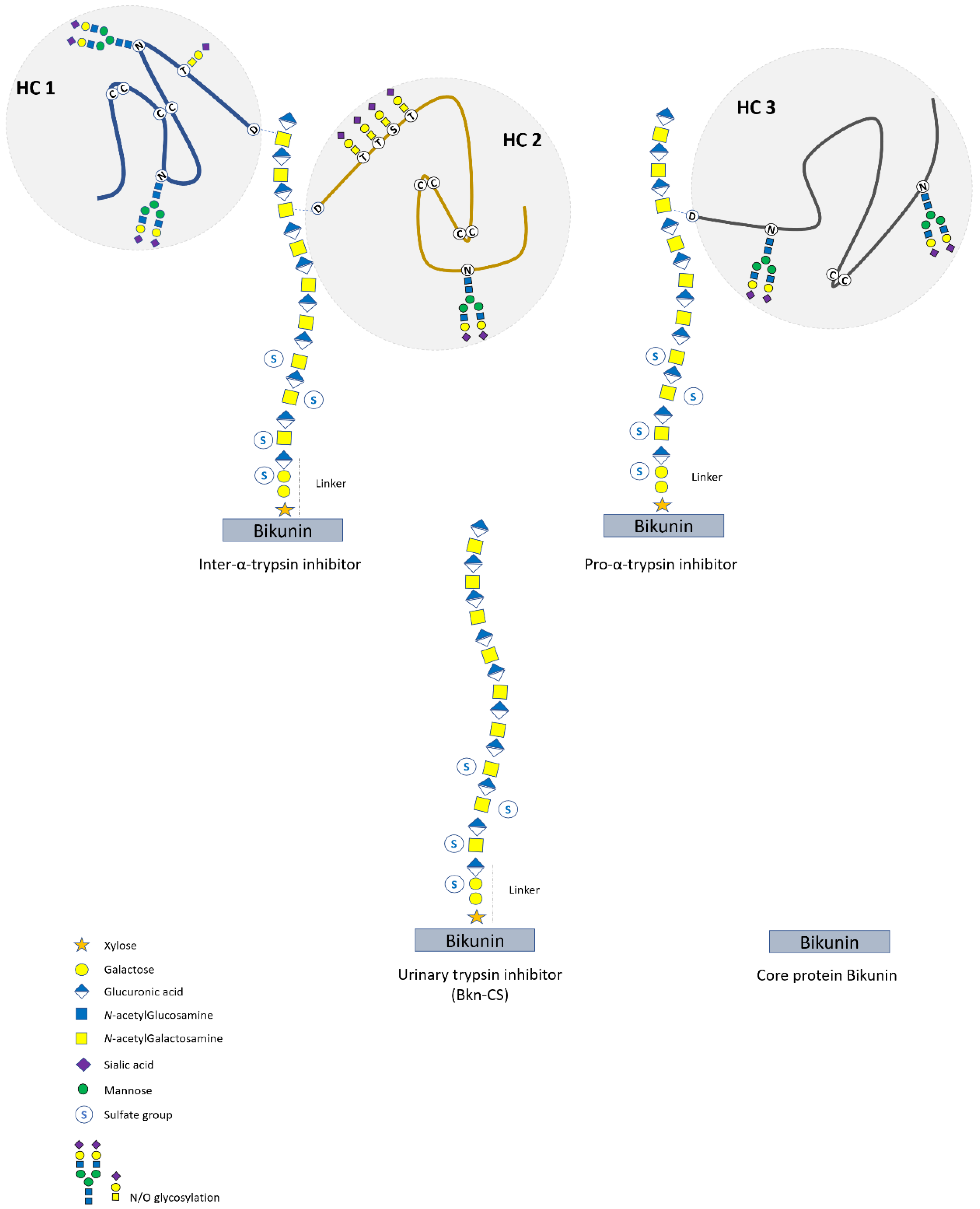
6. Bikunin Proteoglycan Isoforms
7. Serum Bikunin Analyses in PG-IMD
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2-DE | two-dimensional electrophoresis |
| AMP | adenosine monophosphate |
| Bkn | bikunin |
| B3GALT6 | β-3 galactosyltransferase 6 |
| B3GAT3 | β-3 glucuronyltransferase 3 |
| B4GALT7 | β-4 galactosyltransferase 7 |
| CANT1 | calcium activated nucleotidase 1 |
| CHST | chondroitin sulfotransferase |
| CHSY1 | chondroitin synthase 1 |
| COG | conserved oligomeric Golgi |
| COP | coated protein complex |
| CS | chondroitin sulfate |
| CSGALNACT | chondroitin sulfate GalNAc transferase |
| D4ST | dermatan-4-sulfotransferase |
| DS | dermatan sulfate |
| DSE | dermatan sulfate epimerase |
| ECM | extracellular matrix |
| ER | endoplasmic reticulum |
| EXT | exostosin |
| EXTL | exostosin-like |
| FAM20B | family with sequence similarity member 20B |
| GAG | glycosaminoglycan |
| Gal | galactose |
| GalNAc | N-acetygalactosamine |
| GlcA | glucuronic acid |
| GlcN | glucosamine |
| GlcNAc | N-acetylglucosamine |
| GORAB | golgin Rab6-interacting protein |
| HC protein | ‘heavy chain’ protein |
| HMES | hereditary multiple exostosis syndrome |
| HPLC | high-performance liquid chromatography |
| HS | heparan sulfate |
| IAIP | inter-α-trypsin inhibitor proteins |
| IdoA | iduronic acid |
| IMPAD1 | inositol monophosphatase domain-containing 1 |
| ITI | inter-α-trypsin inhibitor |
| KS | keratan sulfate |
| MS | mass spectrometry |
| NDST | N-deacetylase/N-sulfotransferases |
| PAP | phospho-adenosine phosphate |
| PAPS | 3′-phosphoadenosine 5′-phosphosulfate |
| PαI | pro-α-trypsin inhibitor |
| PG | proteoglycan |
| PG-IMD | PG inherited metabolic disorders |
| Ser | serine |
| SLC35 | solute carrier 35 |
| SRLP | small leucin-rich proteoglycans |
| TGN | trans-Golgi network |
| TMEM165 | transmembrane protein 165 |
| U2ST | uronyl 2-O-sulfotransferase or uronosyl 2-O-sulfotransferase |
| UTI | urinary trypsin inhibitor |
| UDP/UMP | uridine di/monophosphate |
| Xyl | xylose |
| XYLT | xylosyltransferase |
| XYLP | xylose phosphatase |
References
- Schaefer, L.; Schaefer, R.M. Proteoglycans: From structural compounds to signaling molecules. Cell Tissue Res. 2010, 339, 237–246. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef]
- Bartlett, A.H.; Park, W. Proteoglycans in host-pathogen interactions: Molecular mechanisms and therapeutic implications. Expert Rev. Mol. Med. 2010, 12, e5. [Google Scholar] [CrossRef] [Green Version]
- Mulloy, B.; Rider, C.C. Cytokines and proteoglycans: An introductory overview. Biochem. Soc. Trans. 2010, 34, 409–413. [Google Scholar] [CrossRef]
- Mizumoto, S.; Yamada, S.; Sugahara, K. Molecular interactions between chondroitin–dermatan sulfate and growth factors/receptors/matrix proteins. Curr. Opin. Struct. Biol. 2015, 34, 35–42. [Google Scholar] [CrossRef]
- Dubail, J.; Cormier-Daire, V. Chondrodysplasias with Multiple Dislocations Caused by Defects in Glycosaminoglycan Synthesis. Front. Genet. 2021, 12, 642097. [Google Scholar] [CrossRef] [PubMed]
- Sasarman, F.; Maftei, C.; Campeau, M.; Brunel-Guitton, C.; Mitchell, G.A. Allard, Biosynthesis of glycosaminoglycans: Associated disorders and biochemical tests. J. Inherit. Metab. Dis. 2016, 39, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Paganini, C.; Costantini, R.; Superti-Furga, A.; Rossi, A. Bone and connective tissue disorders caused by defects in glycosaminoglycan biosynthesis: A panoramic view. FEBS J. 2019, 286, 3008–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muenzer, J. Overview of the mucopolysaccharidoses. Rheumatology 2011, 50, v4–v12. [Google Scholar] [CrossRef] [Green Version]
- Haouari, W.; Dubail, J.; Lounis-Ouaras, S.; Prada, P.; Bennani, R.; Roseau, C.; Huber, C.; Afenjar, A.; Colin, E.; Vuillaumier-Barrot, S.; et al. Serum bikunin isoforms in congenital disorders of glycosylation and linkeropathies. J. Inherit. Metab. Dis. 2020, 43, 1349–1359. [Google Scholar] [CrossRef] [PubMed]
- Prydz, K.; Dalen, K.T. Synthesis and srting of proteoglycans. J. Cell Sci. 2000, 113, 193–205. [Google Scholar] [CrossRef]
- Almeida, R.; Levery, S.B.; Mandel, U.; Kresse, H.; Schwientek, T.; Bennett, E.P.; Clausen, H. Cloning and expression of a proteoglycan UDP-galactose:beta-xylose beta1,4-galactosyltransferase I. A seventh member of the human beta4-galactosyltransferase gene family. J. Biol. Chem. 1999, 274, 26165–26171. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Zhou, D.; Brown, J.R.; Crawford, B.E.; Hennet, T.; Esko, J.D. Biosynthesis of the linkage region of glycosaminoglycans: Cloning and activity of galactosyltransferase II, the sixth member of the beta 1,3-galactosyltransferase family (beta 3GalT6). J. Biol. Chem. 2001, 276, 48189–48195. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, H.; Tone, Y.; Tamura, J.; Neumann, K.W.; Ogawa, T.; Oka, S.; Kawasaki, T.; Sugahara, K. Molecular Cloning and Expression of Glucuronyltransferase I Involved in the Biosynthesis of the Glycosaminoglycan-Protein Linkage Region of Proteoglycans. J. Biol. Chem. 1998, 273, 6615–6618. [Google Scholar] [CrossRef] [Green Version]
- Koike, T.; Izumikawa, T.; Tamura, J.I.; Kitagawa, H. FAM20B is a kinase that phosphorylates xylose in the glycosaminoglycan-protein linkage region. Biochem. J. 2009, 421, 157–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koike, T.; Izumikawa, T.; Sato, B.; Kitagawa, H. Identification of Phosphatase That Dephosphorylates Xylose in the Glycosaminoglycan-Protein Linkage Region of Proteoglycans. J. Biol. Chem. 2014, 289, 6695–6708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulberti, S.; Jacquinet, J.-C.; Chabel, M.; Ramalanjaona, N.; Magdalou, J.; Netter, P.; Coughtrie, M.W.H.; Ouzzine, M.; Fournel-Gigleux, S. Chondroitin sulfate N-acetylgalactosaminyltransferase-1 (CSGalNAcT-1) involved in chondroitin sulfate initiation: Impact of sulfation on activity and specificity. Glycobiology 2012, 22, 561–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitagawa, H.; Tanaka, Y.; Tsuchida, K.; Goto, F.; Ogawa, T.; Lidholt, K.; Lindahl, U.; Sugahara, K. N-acetylgalactosamine (GalNAc) transfer to the common carbohydrate-protein linkage region of sulfated glycosaminoglycans. Identification of UDP-GalNAc:chondro-oligosaccharide alpha-N-acetylgalactosaminyltransferase in fetal bovine serum. J. Biol. Chem. 1995, 270, 22190–22195. [Google Scholar] [CrossRef] [Green Version]
- Uyama, T.; Kitagawa, H.; Tamura, J.; Sugahara, K. Molecular cloning and expression of human chondroitin N-acetylgalactosaminyltransferase: The key enzyme for chain initiation and elongation of chondroitin/dermatan sulfate on the protein linkage region tetrasaccharide shared by heparin/heparan sulfate. J. Biol. Chem. 2002, 277, 8841–8846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitagawa, H.; Uyama, T.; Sugahara, K. Molecular cloning and expression of a human chondroitin synthase. J. Biol. Chem. 2001, 276, 38721–38726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitagawa, H.; Izumikawa, T.; Uyama, T.; Sugahara, K. Molecular Cloning of a Chondroitin Polymerizing Factor That Cooperates with Chondroitin Synthase for Chondroitin Polymerization. J. Biol. Chem. 2003, 278, 23666–23671. [Google Scholar] [CrossRef] [Green Version]
- Izumikawa, T.; Uyama, T.; Okuura, Y.; Sugahara, K.; Kitagawa, H. Involvement of chondroitin sulfate synthase-3 (chondroitin synthase-2) in chondroitin polymerization through its interaction with chondroitin synthase-1 or chondroitin-polymerizing factor. Biochem. J. 2007, 403, 545–552. [Google Scholar] [CrossRef]
- Izumikawa, T.; Koike, T.; Shiozawa, S.; Sugahara, K.; Tamura, J.; Kitagawa, H. Identification of chondroitin sulfate glucuronyltransferase as chondroitin synthase-3 involved in chondroitin polymerization: Chondroitin polymerization is achieved by multiple enzyme complexes consisting of chondroitin synthase family members. J. Biol. Chem. 2008, 283, 11396–11406. [Google Scholar] [CrossRef] [Green Version]
- Lind, T.; Tufaro, F.; McCormick, C.; Lindahl, U.; Lidholt, K. The putative tumor suppressors EXT1 and EXT2 are glycosyltransferases required for the biosynthesis of heparan sulfate. J. Biol. Chem. 1998, 273, 26265–26268. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.T.; Kitagawa, H.; Tamura, J.; Saito, T.; Kusche-Gullberg, M.; Lindahl, U.; Sugahara, K. Human tumor suppressor EXT gene family members EXTL1 and EXTL3 encode alpha 1,4- N-acetylglucosaminyltransferases that likely are involved in heparan sulfate/ heparin biosynthesis. Proc. Natl. Acad. Sci. USA 2001, 98, 7176–7181. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, H.; Shimakawa, H.; Sugahara, K. The tumor suppressor EXT-like gene EXTL2 encodes an alpha1, 4-N-acetylhexosaminyltransferase that transfers N-acetylgalactosamine and N-acetylglucosamine to the common glycosaminoglycan-protein linkage region. The key enzyme for the chain initiation of heparan sulfate. J. Biol. Chem. 1999, 274, 13933–13937. [Google Scholar]
- Seko, A.; Dohmae, N.; Takio, K.; Yamashita, K. Beta 1,4-galactosyltransferase (beta 4GalT)-IV is specific for GlcNAc 6-O-sulfate. Beta 4GalT-IV acts on keratan sulfate-related glycans and a precursor glycan of 6-sulfosialyl-Lewis X. J. Biol. Chem. 2003, 278, 9150–9158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seko, A.; Yamashita, K. beta1,3-N-Acetylglucosaminyltransferase-7 (beta3Gn-T7) acts efficiently on keratan sulfate-related glycans. FEBS Lett. 2004, 556, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Kitayama, K.; Hayashida, Y.; Nishida, K.; Akama, T.O. Enzymes responsible for synthesis of corneal keratan sulfate glycosaminoglycans. J. Biol. Chem. 2007, 282, 30085–30096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, S.; Mita, S.; Matsubara, T.; Fukuta, M.; Habuchi, H.; Kimata, K.; Habuchi, O. Molecular cloning and expression of chondroitin 4-sulfotransferase. J. Biol. Chem. 2000, 275, 8975–8981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiraoka, N.; Nakagawa, H.; Ong, E.; Akama, T.O.; Fukuda, M.N.; Fukuda, M. Molecular cloning and expression of two distinct human chondroitin 4-O-sulfotransferases that belong to the HNK-1 sulfotransferase gene family. J. Biol. Chem. 2000, 275, 20188–20196. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.G.; Evers, M.R.; Xia, G.; Baenziger, J.U.; Schachner, M. Molecular cloning and characterization of chondroitin-4-O-sulfotransferase-3. A novel member of the HNK-1 family of sulfotransferases. J. Biol. Chem. 2002, 277, 34766–34772. [Google Scholar] [CrossRef] [Green Version]
- Fukuta, M.; Uchimura, K.; Nakashima, K.; Kato, M.; Kimata, K.; Shinomura, T.; Habuchi, O. Molecular cloning and expression of chick chondrocyte chondroitin 6-sulfotransferase. J. Biol. Chem. 1995, 270, 18575–18580. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Sugumaran, G.; Liu, J.; Shworak, N.W.; Silbert, J.E.; Rosenberg, R.D. Molecular cloning and characterization of a human uronyl 2-sulfotransferase that sulfates iduronyl and glucuronyl residues in dermatan/chondroitin sulfate. J. Biol. Chem. 1999, 274, 10474–10480. [Google Scholar] [CrossRef] [Green Version]
- Maccarana, M.; Olander, B.; Malmström, J.; Tiedemann, K.; Aebersold, R.; Lindahl, U.; Li, J.-P.; Malmström, A. Biosynthesis of dermatan sulfate: Chondroitin-glucuronate C5-epimerase is identical to SART2. J. Biol. Chem. 2006, 281, 11560–11568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco, B.; Malmström, A.; Maccarana, M. Two dermatan sulfate epimerases form iduronic acid domains in dermatan sulfate. J. Biol. Chem. 2009, 284, 9788–9795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evers, M.R.; Xia, G.; Kang, H.G.; Schachner, M.; Baenziger, J.U. Molecular cloning and characterization of a dermatan-specific N-acetylgalactosamine 4-O-sulfotransferase. J. Biol. Chem. 2001, 276, 36344–36353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, Y.; Orellana, A.; Gil, G.; Hirschberg, C.B. Molecular cloning and expression of rat liver N-heparan sulfate sulfotransferase. J. Biol. Chem. 1992, 267, 15744–15750. [Google Scholar] [CrossRef]
- Eriksson, I.; Sandbäck, D.; Ek, B.; Lindahl, U.; Kjellén, L. cDNA cloning and sequencing of mouse mastocytoma glucosaminyl N-deacetylase/N-sulfotransferase, an enzyme involved in the biosynthesis of heparin. J. Biol. Chem. 1994, 269, 10438–10443. [Google Scholar] [CrossRef]
- Habuchi, H.; Tanaka, M.; Habuchi, O.; Yoshida, K.; Suzuki, H.; Ban, K.; Kimata, K. The occurrence of three isoforms of heparan sulfate 6-O-sulfotransferase having different specificities for hexuronic acid adjacent to the targeted N-sulfoglucosamine. J. Biol. Chem. 2000, 275, 2859–2868. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Hagner-McWhirter, A.; Kjellén, L.; Palgi, J.; Jalkanen, M.; Lindahl, U. Biosynthesis of heparin/heparan sulfate. cDNA cloning and expression of D-glucuronyl C5-epimerase from bovine lung. J. Biol. Chem. 1997, 272, 28158–28163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuta, M.; Inazawa, J.; Torii, T.; Tsuzuki, K.; Shimada, E.; Habuchi, O. Molecular cloning and characterization of human keratan sulfate Gal-6-sulfotransferase. J. Biol. Chem. 1997, 272, 32321–32328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akama, T.O.; Nakayama, J.; Nishida, K.; Hiraoka, N.; Suzuki, M.; McAuliffe, J.; Hindsgaul, O.; Fukuda, M.; Fukuda, M.N. Human corneal GlcNac 6-O-sulfotransferase and mouse intestinal GlcNac 6-O-sulfotransferase both produce keratan sulfate. J. Biol. Chem. 2001, 276, 16271–16278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akama, T.O.; Misra, A.K.; Hindsgaul, O.; Fukuda, M.N. Enzymatic synthesis in vitro of the disulfated disaccharide unit of corneal keratan sulfate. J. Biol. Chem. 2002, 277, 42505–42513. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, J.; Noborn, F.; Gomez Toledo, A.; Nasir, W.; Sihlbom, C.; Larson, G. Characterization of Glycan Structures of Chondroitin Sulfate-Glycopeptides Facilitated by Sodium Ion-Pairing and Positive Mode LC-MS/MS. J. Am. Soc. Mass Spectrom. 2017, 28, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Opat, A.S.; Van Vliet, C.; Gleeson, A. Trafficking and localisation of resident Golgi glycosylation enzymes. Biochimie 2001, 83, 763–773. [Google Scholar] [CrossRef]
- Blackburn, J.B.; D’Souza, Z.; Lupashin, V.V. Maintaining order: COG complex controls Golgi trafficking, processing, and sorting. FEBS Lett. 2019, 593, 2466–2487. [Google Scholar] [CrossRef] [Green Version]
- Yarema, K.J.; Bertozzi, C.R. Characterizing glycosylation pathways. Genome Biol. 2001, 2, REVIEWS0004. [Google Scholar] [CrossRef]
- Kleczkowski, L.A.; Decker, D.; Wilczynska, M. UDP-Sugar Pyrophosphorylase: A New Old Mechanism for Sugar Activation. Plant Physiol. 2011, 156, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Hirschberg, C.B. My journey in the discovery of nucleotide sugar transporters of the Golgi apparatus. J. Biol. Chem. 2018, 293, 12653–12662. [Google Scholar] [CrossRef] [Green Version]
- Song, Z. Roles of the nucleotide sugar transporters (SLC35 family) in health and disease. Mol. Asp. Med. 2013, 34, 590–600. [Google Scholar] [CrossRef]
- Paganini, C.; Monti, L.; Costantini, R.; Besio, R.; Lecci, S.; Biggiogera, M.; Tian, K.; Schwartz, J.-M.; Huber, C.; Cormier-Daire, V.; et al. Calcium activated nucleotidase 1 (CANT1) is critical for glycosaminoglycan biosynthesis in cartilage and endochondral ossification. Matrix Biol. 2019, 81, 70. [Google Scholar] [CrossRef]
- Fuda, H.; Shimizu, C.; Lee, Y.C.; Akita, H.; Strott, C.A. Characterization and expression of human bifunctional 3′-phosphoadenosine 5′-phosphosulphate synthase isoforms. Biochem. J. 2002, 365, 497–504. [Google Scholar] [CrossRef] [Green Version]
- Kamiyama, S.; Suda, T.; Ueda, R.; Suzuki, M.; Okubo, R.; Kikuchi, N.; Chiba, Y.; Goto, S.; Toyoda, H.; Saigo, K.; et al. Molecular cloning and identification of 3’-phosphoadenosine 5’-phosphosulfate transporter. J. Biol. Chem. 2003, 278, 25958–25963. [Google Scholar] [CrossRef] [Green Version]
- Kamiyama, S.; Sasaki, N.; Goda, E.; Ui-Tei, K.; Saigo, K.; Narimatsu, H.; Jigami, Y.; Kannagi, R.; Irimura, T.; Nishihara, S. Molecular Cloning and Characterization of a Novel 3′-Phosphoadenosine 5′-Phosphosulfate Transporter, PAPST2. J. Biol. Chem. 2006, 281, 10945–10953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vissers, L.E.; Lausch, E.; Unger, S.; Campos-Xavier, A.B.; Gilissen, C.; Rossi, A.; Del Rosario, M.; Venselaar, H.; Knoll, U.; Nampoothiri, S.; et al. Chondrodysplasia and Abnormal Joint Development Associated with Mutations in IMPAD1, Encoding the Golgi-Resident Nucleotide Phosphatase, Gpapam. J. Hum. Genet. 2011, 88, 608–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolset, S.O.; Tveit, H. Serglycin—Structure and biology. Cell. Mol. Life Sci. 2008, 65, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- De Rossi, G.; Evans, A.R.; Kay, E.; Woodfin, A.; McKay, T.R.; Nourshargh, S.; Whiteford, J.R. Shed syndecan-2 inhibits angiogenesis. J. Cell Sci. 2014, 127, 4788–4799. [Google Scholar] [CrossRef] [Green Version]
- Xian, X.; Gopal, S.; Couchman, J.R. Syndecans as receptors and organizers of the extracellular matrix. Cell Tissue Res. 2010, 339, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Chung, H.; Jung, H.; Couchman, J.R.; Oh, E.-S. Syndecans as cell surface receptors: Unique structure equates with functional diversity. Matrix Biol. 2011, 30, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Lord, M.S.; Tang, F.; Rnjak-Kovacina, J.; Smith, J.G.W.; Melrose, J.; Whitelock, J.M. The multifaceted roles of perlecan in fibrosis. Matrix Biol. 2018, 68–69, 150–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Hu, W.; Guo, F.; Zhang, W.; Wang, J.; Chen, A. Glycosaminoglycan chains of biglycan promote bone morphogenetic protein-4-induced osteoblast differentiation. Int. J. Mol. Med. 2012, 30, 1075–1080. [Google Scholar] [CrossRef] [Green Version]
- Rühland, C.; Schönherr, E.; Robenek, H.; Hansen, U.; Iozzo, R.V.; Bruckner, P.; Seidler, D.G. The glycosaminoglycan chain of decorin plays an important role in collagen fibril formation at the early stages of fibrillogenesis. FEBS J. 2007, 274, 4246–4255. [Google Scholar] [CrossRef]
- Kali, A.; Shetty, K.S.R. Endocan: A novel circulating proteoglycan. Indian J. Pharm. 2014, 46, 579–583. [Google Scholar] [CrossRef] [Green Version]
- Bui, C.; Huber, C.; Tuysuz, B.; Alanay, Y.; Bole-Feysot, C.; Leroy, J.G.; Mortier, G.; Nitschke, P.; Munnich, A.; Cormier-Daire, V. XYLT1 Mutations in Desbuquois Dysplasia Type 2. Am. J. Hum. Genet. 2014, 94, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munns, C.F.; Fahiminiya, S.; Poudel, N.; Munteanu, M.C.; Majewski, J.; Sillence, D.O.; Metcalf, J.P.; Biggin, A.; Glorieux, F.; Fassier, F.; et al. Homozygosity for frameshift mutations in XYLT2 result in a spondylo-ocular syndrome with bone fragility, cataracts, and hearing defects. Am. J. Hum. Genet. 2015, 96, 971–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malfait, F.; Kariminejad, A.; Van Damme, T.; Gauche, C.; Syx, D.; Merhi-Soussi, F.; Gulberti, S.; Symoens, S.; Vanhauwaert, S.; Willaert, A.; et al. Defective Initiation of Glycosaminoglycan Synthesis due to B3GALT6 Mutations Causes a Pleiotropic Ehlers-Danlos-Syndrome-like Connective Tissue Disorder. Am. J. Hum. Genet. 2013, 92, 935–945. [Google Scholar] [CrossRef] [Green Version]
- Baasanjav, S.; Al-Gazali, L.; Hashiguchi, T.; Mizumoto, S.; Fischer, B.; Horn, D.; Seelow, D.; Ali, B.R.; Aziz, S.A.A.; Langer, R.; et al. Faulty Initiation of Proteoglycan Synthesis Causes Cardiac and Joint Defects. Am. J. Hum. Genet. 2011, 89, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.; Lüdecke, H.J.; Lindow, S.; Horton, W.A.; Lee, B.; Wagner, M.J.; Horsthemke, B.; Wells, D.E. Cloning of the putative tumour suppressor gene for hereditary multiple exostoses (EXT1). Nat. Genet. 1995, 11, 137–143. [Google Scholar] [CrossRef]
- Stickens, D.; Clines, G.; Burbee, D.; Ramos, P.; Thomas, S.; Hogue, D.; Hecht, J.T.; Lovett, M.; Evans, G.A. The EXT2 multiple exostoses gene defines a family of putative tumour suppressor genes. Nat. Genet. 1996, 14, 25–32. [Google Scholar] [CrossRef]
- Soares da Costa, D.; Reis, R.L.; Pashkuleva, I. Sulfation of Glycosaminoglycans and Its Implications in Human Health and Disorders. Annu. Rev. Biomed. Eng. 2017, 19, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Péanne, R.; de Lonlay, P.; Foulquier, F.; Kornak, U.; Lefeber, D.J.; Morava, E.; Pérez, B.; Seta, N.; Thiel, C.; Van Schaftingen, E.; et al. Congenital disorders of glycosylation (CDG): Quo Vadis? Eur. J. Med. Genet. 2018, 61, 643–663. [Google Scholar] [CrossRef]
- Toma, L.; Pinhal, M.A.S.; Dietrich, C.P.; Nader, H.B.; Hirschberg, C.B. Transport of UDP-Galactose into the Golgi Lumen Regulates the Biosynthesis of Proteoglycans. J. Biol. Chem. 1996, 271, 3897–3901. [Google Scholar] [CrossRef] [Green Version]
- Maszczak-Seneczko, D.; Olczak, T.; Wunderlich, L.; Olczak, M. Comparative analysis of involvement of UGT1 and UGT2 splice variants of UDP-galactose transporter in glycosylation of macromolecules in MDCK and CHO cell lines. Glycoconj. J. 2011, 28, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, C.R.; Xia, Z.-J.; Clément, A.; Parry, D.A.; Davids, M.; Taylan, F.; Sharma, P.; Turgeon, C.T.; Blanco-Sánchez, B.; Ng, B.G.; et al. A Recurrent De Novo Heterozygous COG4 Substitution Leads to Saul-Wilson Syndrome, Disrupted Vesicular Trafficking, and Altered Proteoglycan Glycosylation. Am. J. Hum. Genet. 2018, 103, 553–567. [Google Scholar] [CrossRef]
- Hennies, H.C.; Kornak, U.; Zhang, H.; Egerer, J.; Zhang, X.; Seifert, W.; Kühnisch, J.; Budde, B.; Nätebus, M.; Brancati, F.; et al. Gerodermia osteodysplastica is caused by mutations in SCYL1BP1, a Rab-6 interacting golgin. Nat. Genet. 2008, 40, 1410–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bammens, R.; Mehta, N.; Race, V.; Foulquier, F.; Jaeken, J.; Tiemeyer, M.; Steet, R.; Matthijs, G.; Flanagan-Steet, H. Abnormal cartilage development and altered N-glycosylation in Tmem165-deficient zebrafish mirrors the phenotypes associated with TMEM165-CDG. Glycobiology 2015, 25, 669–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubail, J.; Huber, C.; Chantepie, S.; Sonntag, S.; Tüysüz, B.; Mihci, E.; Gordon, C.T.; Steichen-Gersdorf, E.; Amiel, J.; Nur, B.; et al. SLC10A7 mutations cause a skeletal dysplasia with amelogenesis imperfecta mediated by GAG biosynthesis defects. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hengel, H.; Bosso-Lefèvre, C.; Grady, G.; Szenker-Ravi, E.; Li, H.; Pierce, S.; Lebigot, É.; Tan, T.-T.; Eio, M.Y.; Narayanan, G.; et al. Loss-of-function mutations in UDP-Glucose 6-Dehydrogenase cause recessive developmental epileptic encephalopathy. Nat. Commun. 2020, 11, 595. [Google Scholar] [CrossRef] [Green Version]
- Nizon, M.; Huber, C.; De Leonardis, F.; Merrina, R.; Forlino, A.; Fradin, M.; Tuysuz, B.; Abu-Libdeh, B.Y.; Alanay, Y.; Albrecht, B.; et al. Further Delineation of CANT1 Phenotypic Spectrum and Demonstration of Its Role in Proteoglycan Synthesis. Hum. Mutat. 2012, 33, 1261–1266. [Google Scholar] [CrossRef] [Green Version]
- Mizumoto, S.; Janecke, A.R.; Sadeghpour, A.; Povysil, G.; McDonald, M.T.; Unger, S.; Greber-Platzer, S.; Deak, K.L.; Katsanis, N.; Superti-Furga, A.; et al. CSGALNACT1-congenital disorder of glycosylation: A mild skeletal dysplasia with advanced bone age. Hum. Mutat. 2020, 41, 655–667. [Google Scholar] [CrossRef] [Green Version]
- Müller, T.; Mizumoto, S.; Suresh, I.; Komatsu, Y.; Vodopiutz, J.; Dundar, M.; Straub, V.; Lingenhel, A.; Melmer, A.; Lechner, S.; et al. Loss of dermatan sulfate epimerase (DSE) function results in musculocontractural Ehlers–Danlos syndrome. Hum. Mol. Genet. 2013, 22, 3761–3772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dündar, M.; Müller, T.; Zhang, Q.; Pan, J.; Steinmann, B.; Vodopiutz, J.; Gruber, R.; Sonoda, T.; Krabichler, B.; Utermann, G.; et al. Loss of Dermatan-4-Sulfotransferase 1 Function Results in Adducted Thumb-Clubfoot Syndrome. Am. J. Hum. Genet. 2009, 85, 873–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidler, D.G.; Faiyaz-Ul-Haque, M.; Hansen, U.; Yip, G.W.; Zaidi, S.H.E.; Teebi, A.S.; Kiesel, L.; Götte, M. Defective glycosylation of decorin and biglycan, altered collagen structure, and abnormal phenotype of the skin fibroblasts of an Ehlers-Danlos syndrome patient carrying the novel Arg270Cys substitution in galactosyltransferase I (beta4GalT-7). J. Mol. Med. 2006, 84, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Edvardson, S.; Ashikov, A.; Jalas, C.; Sturiale, L.; Shaag, A.; Fedick, A.; Treff, N.R.; Garozzo, D.; Gerardy-Schahn, R.; Elpeleg, O. Mutations in SLC35A3 cause autism spectrum disorder, epilepsy and arthrogryposis. J. Med. Genet. 2013, 50, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Anower-E-Khuda, M.F.; Matsumoto, K.; Habuchi, H.; Morita, H.; Yokochi, T.; Shimizu, K.; Kimata, K. Glycosaminoglycans in the blood of hereditary multiple exostoses patients: Half reduction of heparan sulfate to chondroitin sulfate ratio and the possible diagnostic application. Glycobiology 2013, 23, 865–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oud, M.M.; Tuijnenburg, P.; Hempel, M.; van Vlies, N.; Ren, Z.; Ferdinandusse, S.; Jansen, M.H.; Santer, R.; Johannsen, J.; Bacchelli, C.; et al. Mutations in EXTL3 Cause Neuro-immuno-skeletal Dysplasia Syndrome. Am. J. Hum. Genet. 2017, 100, 281–296. [Google Scholar] [CrossRef] [Green Version]
- Thiele, H. Loss of chondroitin 6-O-sulfotransferase-1 function results in severe human chondrodysplasia with progressive spinal involvement. Proc. Natl. Acad. Sci. USA 2004, 101, 10155–10160. [Google Scholar] [CrossRef] [Green Version]
- Kubaski, F.; de Oliveira Poswar, F.; Michelin-Tirelli, K.; Burin, M.G.; Rojas-Málaga, D.; Brusius-Facchin, A.C.; Leistner-Segal, S.; Giugliani, R. Diagnosis of Mucopolysaccharidoses. Diagnostics 2020, 10, 172. [Google Scholar] [CrossRef] [Green Version]
- Bruneel, A.; Dubail, J.; Roseau, C.; Prada, P.; Haouari, W.; Huber, C.; Dupré, T.; Poüs, C.; Cormier-Daire, V.; Seta, N. Serum bikunin is a biomarker of linkeropathies. Clin. Chim. Acta 2018, 485, 178–180. [Google Scholar] [CrossRef]
- Enghild, J.J.; Thøgersen, I.B.; Cheng, F.; Fransson, L.A.; Roepstorff, P.; Rahbek-Nielsen, H. Organization of the inter-alpha-inhibitor heavy chains on the chondroitin sulfate originating from Ser(10) of bikunin: Posttranslational modification of IalphaI-derived bikunin. Biochemistry 1999, 38, 11804–11813. [Google Scholar] [CrossRef]
- Zhuo, L.; Hascall, V.C.; Kimata, K. Inter-α-trypsin Inhibitor, a Covalent Protein-Glycosaminoglycan-Protein Complex. J. Biol. Chem. 2004, 279, 38079–38082. [Google Scholar] [CrossRef] [Green Version]
- Pugia, M.J.; Jortani, S.A.; Basu, M.; Sommer, R.; Kuo, H.-H.; Murphy, S.; Williamson, D.; Vranish, J.; Boyle, P.J.; Budzinski, D.; et al. Immunological evaluation of urinary trypsin inhibitors in blood and urine: Role of N- & O-linked glycoproteins. Glycoconj. J. 2007, 24, 5–15. [Google Scholar] [PubMed]
- Lepedda, A.J.; Nieddu, G.; Rocchiccioli, S.; Fresu, P.; De Muro, P.; Formato, M. Development of a method for urine bikunin/urinary trypsin inhibitor (UTI) quantitation and structural characterization: Application to type 1 and type 2 diabetes. Electrophoresis 2013, 34, 3227–3233. [Google Scholar] [CrossRef]
- Lim, Y.-P.; Bendelja, K.; Opal, S.M.; Siryaporn, E.; Hixson, D.C.; Palardy, J.E. Correlation between mortality and the levels of inter-alpha inhibitors in the plasma of patients with severe sepsis. J. Infect. Dis. 2003, 188, 919–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamm, A.; Veeck, J.; Bektas, N.; Wild, P.J.; Hartmann, A.; Heindrichs, U.; Kristiansen, G.; Werbowetski-Ogilvie, T.; Del Maestro, R.; Knuechel, R.; et al. Frequent expression loss of Inter-alpha-trypsin inhibitor heavy chain (ITIH) genes in multiple human solid tumors: A systematic expression analysis. BMC Cancer 2008, 8, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, D.C.; Langford-Smith, A.W.W.; Birchenough, H.L.; Jowitt, T.A.; Kielty, C.M.; Enghild, J.J.; Baldock, C.; Milner, C.M.; Day, A.J. Inter-α-inhibitor heavy chain-1 has an integrin-like 3D structure mediating immune regulatory activities and matrix stabilization during ovulation. J. Biol. Chem. 2020, 295, 5278–5291. [Google Scholar] [CrossRef] [Green Version]
- Yingsung, W.; Zhuo, L.; Morgelin, M.; Yoneda, M.; Kida, D.; Watanabe, H.; Ishiguro, N.; Iwata, H.; Kimata, K. Molecular heterogeneity of the SHAP-hyaluronan complex. Isolation and characterization of the complex in synovial fluid from patients with rheumatoid arthritis. J. Biol. Chem. 2003, 278, 32710–32718. [Google Scholar] [CrossRef] [Green Version]
- McDonald, B. Interaction of CD44 and hyaluronan is the dominant mechanism for neutrophil sequestration in inflamed liver sinusoids. J. Exp. Med. 2008, 205, 915–927. [Google Scholar] [CrossRef] [Green Version]
- Lepedda, A.J.; De Muro, P.; Capobianco, G.; Formato, M. Role of the small proteoglycan bikunin in human reproduction. Hormones 2019, 19, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, H.G.; Hua, J.C.; Poppers, D.M.; Naime, D.; Vilcek, J.; Cronstein, B.N. TNF/IL-1-inducible protein TSG-6 potentiates plasmin inhibition by inter-alpha-inhibitor and exerts a strong anti-inflammatory effect in vivo. J. Immunol. 1996, 156, 1609–1615. [Google Scholar] [PubMed]
- Adair, J.E.; Stober, V.; Sobhany, M.; Zhuo, L.; Roberts, J.D.; Negishi, M.; Kimata, K.; Garantziotis, S. Inter-α-trypsin Inhibitor Promotes Bronchial Epithelial Repair after Injury through Vitronectin Binding. J. Biol. Chem. 2009, 284, 16922–16930. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haouari, W.; Dubail, J.; Poüs, C.; Cormier-Daire, V.; Bruneel, A. Inherited Proteoglycan Biosynthesis Defects—Current Laboratory Tools and Bikunin as a Promising Blood Biomarker. Genes 2021, 12, 1654. https://doi.org/10.3390/genes12111654
Haouari W, Dubail J, Poüs C, Cormier-Daire V, Bruneel A. Inherited Proteoglycan Biosynthesis Defects—Current Laboratory Tools and Bikunin as a Promising Blood Biomarker. Genes. 2021; 12(11):1654. https://doi.org/10.3390/genes12111654
Chicago/Turabian StyleHaouari, Walid, Johanne Dubail, Christian Poüs, Valérie Cormier-Daire, and Arnaud Bruneel. 2021. "Inherited Proteoglycan Biosynthesis Defects—Current Laboratory Tools and Bikunin as a Promising Blood Biomarker" Genes 12, no. 11: 1654. https://doi.org/10.3390/genes12111654
APA StyleHaouari, W., Dubail, J., Poüs, C., Cormier-Daire, V., & Bruneel, A. (2021). Inherited Proteoglycan Biosynthesis Defects—Current Laboratory Tools and Bikunin as a Promising Blood Biomarker. Genes, 12(11), 1654. https://doi.org/10.3390/genes12111654