
Beyond Trinucleotide Repeat Expansion in Fragile X Syndrome: Rare Coding and Noncoding Variants in FMR1 and Associated Phenotypes


Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. FMR1 CNVs in the Absence of Repeat Expansion
3.2. Mosaicism for CNVs with CGG Repeat Expansions
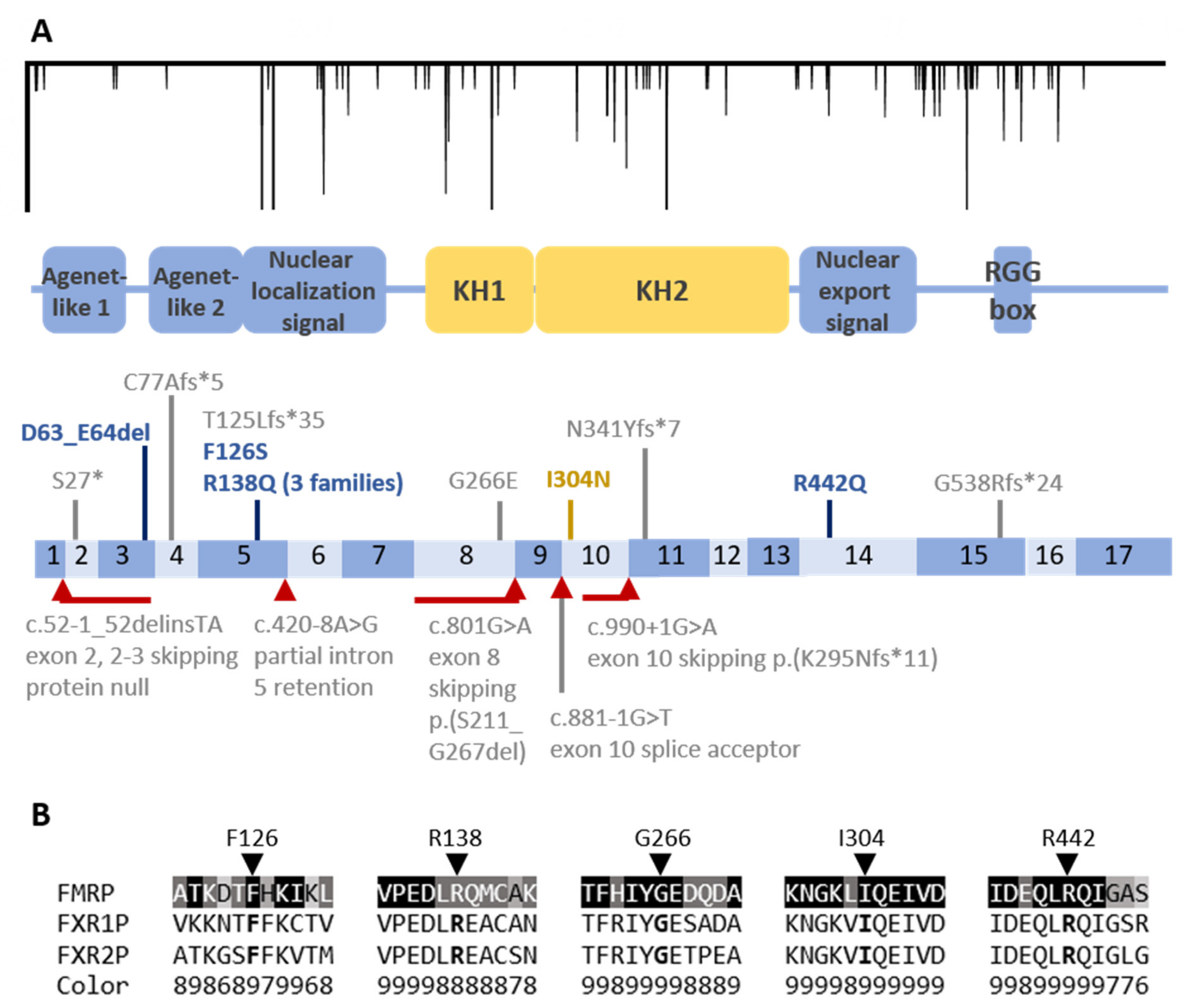
3.3. Coding Region Variants
3.4. Noncoding Small Variants
4. Discussion
4.1. Clinical Presentations of Pathogenic Non-Repeat Expansion Variants
4.2. Noncoding Variants Can Cause Disease, but New Variants Require Functional Confirmation
4.3. Rearrangement Breakpoints around the CGG Repeat
4.4. Detection of FMR1 Variants Other than Repeat Expansions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hersh, J.H.; Saul, R.A.; Committee on, G. Health supervision for children with fragile X syndrome. Pediatrics 2011, 127, 994–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidd, S.A.; Lachiewicz, A.; Barbouth, D.; Blitz, R.K.; Delahunty, C.; McBrien, D.; Visootsak, J.; Berry-Kravis, E. Fragile X syndrome: A review of associated medical problems. Pediatrics 2014, 134, 995–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagerman, R.J.; Jackson, C.; Amiri, K.; Silverman, A.C.; O’Connor, R.; Sobesky, W. Girls with fragile X syndrome: Physical and neurocognitive status and outcome. Pediatrics 1992, 89, 395–400. [Google Scholar] [PubMed]
- Spector, E.; Behlmann, A.; Kronquist, K.; Rose, N.C.; Lyon, E.; Reddi, H.V.; Committee, A.L.Q.A. Laboratory testing for fragile X, 2021 revision: A technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, P. Fragile X-associated tremor/ataxia syndrome (FXTAS): Pathology and mechanisms. Acta Neuropathol. 2013, 126, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Eichler, E.E.; Holden, J.J.; Popovich, B.W.; Reiss, A.L.; Snow, K.; Thibodeau, S.N.; Richards, C.S.; Ward, P.A.; Nelson, D.L. Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nat. Genet. 1994, 8, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Glicksman, A.; Ersalesi, N.; Dobkin, C.; Brown, W.T.; Cao, R.; Blatt, E.; Sah, S.; Latham, G.J.; Hadd, A.G. Fragile X full mutation expansions are inhibited by one or more AGG interruptions in premutation carriers. Genet. Med. 2015, 17, 358–364. [Google Scholar] [CrossRef] [Green Version]
- Wells, R.D. Mutation spectra in fragile X syndrome induced by deletions of CGG*CCG repeats. J. Biol. Chem. 2009, 284, 7407–7411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kononenko, A.V.; Ebersole, T.; Vasquez, K.M.; Mirkin, S.M. Mechanisms of genetic instability caused by (CGG)n repeats in an experimental mammalian system. Nat. Struct. Mol. Biol. 2018, 25, 669–676. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M.; ClinGen Sequence Variant Interpretation Working, G. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef]
- Brandt, T.; Sack, L.M.; Arjona, D.; Tan, D.; Mei, H.; Cui, H.; Gao, H.; Bean, L.J.H.; Ankala, A.; Del Gaudio, D.; et al. Adapting ACMG/AMP sequence variant classification guidelines for single-gene copy number variants. Genet. Med. 2020, 22, 336–344. [Google Scholar] [CrossRef]
- Quan, F.; Zonana, J.; Gunter, K.; Peterson, K.L.; Magenis, R.E.; Popovich, B.W. An atypical case of fragile X syndrome caused by a deletion that includes the FMR1 gene. Am. J. Hum. Genet. 1995, 56, 1042–1051. [Google Scholar] [PubMed]
- Parvari, R.; Mumm, S.; Galil, A.; Manor, E.; Bar-David, Y.; Carmi, R. Deletion of 8.5 Mb, including the FMR1 gene, in a male with the fragile X syndrome phenotype and overgrowth. Am. J. Med. Genet. 1999, 83, 302–307. [Google Scholar] [CrossRef]
- Myers, K.A.; van’t Hof, F.N.G.; Sadleir, L.G.; Legault, G.; Simard-Tremblay, E.; Amor, D.J.; Scheffer, I.E. Fragile Females: Case Series of Epilepsy in Girls With FMR1 Disruption. Pediatrics 2019, 144, e20190599. [Google Scholar] [CrossRef] [PubMed]
- Coffee, B.; Ikeda, M.; Budimirovic, D.B.; Hjelm, L.N.; Kaufmann, W.E.; Warren, S.T. Mosaic FMR1 deletion causes fragile X syndrome and can lead to molecular misdiagnosis: A case report and review of the literature. Am. J. Med. Genet. A 2008, 146A, 1358–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wöhrle, D.; Kotzot, D.; Hirst, M.C.; Manca, A.; Korn, B.; Schmidt, A.; Barbi, G.; Rott, H.D.; Poustka, A.; Davies, K.E.; et al. A microdeletion of less than 250 kb, including the proximal part of the FMR-I gene and the fragile-X site, in a male with the clinical phenotype of fragile-X syndrome. Am. J. Hum. Genet. 1992, 51, 299–306. [Google Scholar] [PubMed]
- Jiraanont, P.; Hagerman, R.J.; Neri, G.; Zollino, M.; Murdolo, M.; Tassone, F. Germinal mosaicism for a deletion of the FMR1 gene leading to fragile X syndrome. Eur. J. Med. Genet. 2016, 59, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Trottier, Y.; Imbert, G.; Poustka, A.; Fryns, J.P.; Mandel, J.L. Male with typical fragile X phenotype is deleted for part of the FMR1 gene and for about 100 kb of upstream region. Am. J. Med. Genet. 1994, 51, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Hirst, M.; Grewal, P.; Flannery, A.; Slatter, R.; Maher, E.; Barton, D.; Fryns, J.P.; Davies, K. Two new cases of FMR1 deletion associated with mental impairment. Am. J. Hum. Genet. 1995, 56, 67–74. [Google Scholar] [PubMed]
- Prior, T.W.; Papp, A.C.; Snyder, P.J.; Sedra, M.S.; Guida, M.; Enrile, B.G. Germline mosaicism at the fragile X locus. Am. J. Med. Genet. 1995, 55, 384–386. [Google Scholar] [CrossRef]
- Griswold, A.J.; Ma, D.; Cukier, H.N.; Nations, L.D.; Schmidt, M.A.; Chung, R.H.; Jaworski, J.M.; Salyakina, D.; Konidari, I.; Whitehead, P.L.; et al. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum. Mol. Genet. 2012, 21, 3513–3523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagamani, S.C.; Erez, A.; Probst, F.J.; Bader, P.; Evans, P.; Baker, L.A.; Fang, P.; Bertin, T.; Hixson, P.; Stankiewicz, P.; et al. Small genomic rearrangements involving FMR1 support the importance of its gene dosage for normal neurocognitive function. Neurogenetics 2012, 13, 333–339. [Google Scholar] [CrossRef]
- Gu, Y.; Lugenbeel, K.A.; Vockley, J.G.; Grody, W.W.; Nelson, D.L. A de novo deletion in FMR1 in a patient with developmental delay. Hum. Mol. Genet. 1994, 3, 1705–1706. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Huang, W.; Xia, Q.; Du, Q.; Wu, L.; Duan, R. Mutational analyses of the FMR1 gene in Chinese pediatric population of fragile x suspects: Low tolerance for point mutation. J. Child. Neurol. 2015, 30, 803–806. [Google Scholar] [CrossRef]
- Luo, S.; Huang, W.; Xia, Q.; Xia, Y.; Du, Q.; Wu, L.; Duan, R. Cryptic FMR1 mosaic deletion in a phenotypically normal mother of a boy with fragile X syndrome: Case report. BMC Med. Genet. 2014, 15, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijer, H.; de Graaff, E.; Merckx, D.M.; Jongbloed, R.J.; de Die-Smulders, C.E.; Engelen, J.J.; Fryns, J.P.; Curfs, P.M.; Oostra, B.A. A deletion of 1.6 kb proximal to the CGG repeat of the FMR1 gene causes the clinical phenotype of the fragile X syndrome. Hum. Mol. Genet. 1994, 3, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Hammond, L.S.; Macias, M.M.; Tarleton, J.C.; Shashidhar Pai, G. Fragile X syndrome and deletions in FMR1: New case and review of the literature. Am. J. Med. Genet. 1997, 72, 430–434. [Google Scholar] [CrossRef]
- Collins, S.C.; Coffee, B.; Benke, P.J.; Berry-Kravis, E.; Gilbert, F.; Oostra, B.; Halley, D.; Zwick, M.E.; Cutler, D.J.; Warren, S.T. Array-based FMR1 sequencing and deletion analysis in patients with a fragile X syndrome-like phenotype. PLoS ONE 2010, 5, e9476. [Google Scholar] [CrossRef] [Green Version]
- Tabolacci, E.; Pietrobono, R.; Maneri, G.; Remondini, L.; Nobile, V.; Della Monica, M.; Pomponi, M.G.; Genuardi, M.; Neri, G.; Chiurazzi, P. Reversion to Normal of FMR1 Expanded Alleles: A Rare Event in Two Independent Fragile X Syndrome Families. Genes 2020, 11, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, C.C.; Lee, C.N.; Wang, Y.C.; Chen, C.L.; Lin, T.K.; Su, Y.N.; Lin, M.W.; Kang, J.; Tai, Y.Y.; Hsu, W.W.; et al. Fragile X syndrome carrier screening in pregnant women in Chinese Han population. Sci. Rep. 2019, 9, 15456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grønskov, K.; Hjalgrim, H.; Bjerager, M.O.; Brøndum-Nielsen, K. Deletion of all CGG repeats plus flanking sequences in FMR1 does not abolish gene expression. Am. J. Hum. Genet. 1997, 61, 961–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erbs, E.; Fenger-Grøn, J.; Jacobsen, C.M.; Lildballe, D.L.; Rasmussen, M. Spontaneous rescue of a FMR1 repeat expansion and review of deletions in the FMR1 non-coding region. Eur. J. Med. Genet. 2021, 64, 104244. [Google Scholar] [CrossRef] [PubMed]
- Redin, C.; Gérard, B.; Lauer, J.; Herenger, Y.; Muller, J.; Quartier, A.; Masurel-Paulet, A.; Willems, M.; Lesca, G.; El-Chehadeh, S.; et al. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J. Med. Genet. 2014, 51, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Quartier, A.; Poquet, H.; Gilbert-Dussardier, B.; Rossi, M.; Casteleyn, A.S.; Portes, V.D.; Feger, C.; Nourisson, E.; Kuentz, P.; Redin, C.; et al. Intragenic FMR1 disease-causing variants: A significant mutational mechanism leading to Fragile-X syndrome. Eur. J. Hum. Genet. 2017, 25, 423–431. [Google Scholar] [CrossRef] [Green Version]
- Viveiros, M.T.; Santos, M.D.; Dos Santos, J.M.; Viveiros, D.M.; Cavalcante, M.R.; Caldas, A.J.; Pimentel, M.M. Screening for fragile X syndrome in males from specialized institutions in the northeast region of Brazil. Genet. Mol. Res. 2015, 14, 6897–6905. [Google Scholar] [CrossRef] [PubMed]
- Vengoechea, J.; Parikh, A.S.; Zhang, S.; Tassone, F. De novo microduplication of the FMR1 gene in a patient with developmental delay, epilepsy and hyperactivity. Eur. J. Hum. Genet. 2012, 20, 1197–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mononen, T.; von Koskull, H.; Airaksinen, R.L.; Juvonen, V. A novel duplication in the FMR1 gene: Implications for molecular analysis in fragile X syndrome and repeat instability. Clin. Genet. 2007, 72, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Snow, K.; Tester, D.J.; Kruckeberg, K.E.; Schaid, D.J.; Thibodeau, S.N. Sequence analysis of the fragile X trinucleotide repeat: Implications for the origin of the fragile X mutation. Hum. Mol. Genet. 1994, 3, 1543–1551. [Google Scholar] [CrossRef] [PubMed]
- Gedeon, A.K.; Meinanen, M.; Ades, L.C.; Kaariainen, H.; Gecz, J.; Baker, E.; Sutherland, G.R.; Mulley, J.C. Overlapping submicroscopic deletions in Xq28 in two unrelated boys with developmental disorders: Identification of a gene near FRAXE. Am. J. Hum. Genet. 1995, 56, 907–914. [Google Scholar] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves, T.F.; dos Santos, J.M.; Gonçalves, A.P.; Tassone, F.; Mendoza-Morales, G.; Ribeiro, M.G.; Kahn, E.; Boy, R.; Pimentel, M.M.; Santos-Rebouças, C.B. Finding FMR1 mosaicism in Fragile X syndrome. Expert Rev. Mol. Diagn. 2016, 16, 501–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Graaff, E.; Rouillard, P.; Willems, P.J.; Smits, A.P.; Rousseau, F.; Oostra, B.A. Hotspot for deletions in the CGG repeat region of FMR1 in fragile X patients. Hum. Mol. Genet. 1995, 4, 45–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, F.; Grompe, M.; Jakobs, P.; Popovich, B.W. Spontaneous deletion in the FMR1 gene in a patient with fragile X syndrome and cherubism. Hum. Mol. Genet. 1995, 4, 1681–1684. [Google Scholar] [CrossRef] [PubMed]
- García Arocena, D.; de Diego, Y.; Oostra, B.A.; Willemsen, R.; Mirta Rodriguez, M. A fragile X case with an amplification/deletion mosaic pattern. Hum. Genet. 2000, 106, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Schmucker, B.; Ballhausen, W.G.; Pfeiffer, R.A. Mosaicism of a microdeletion of 486 bp involving the CGG repeat of the FMR1 gene due to misalignment of GTT tandem repeats at chi-like elements flanking both breakpoints and a full mutation. Hum. Genet. 1996, 98, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Jiraanont, P.; Kumar, M.; Tang, H.T.; Espinal, G.; Hagerman, P.J.; Hagerman, R.J.; Chutabhakdikul, N.; Tassone, F. Size and methylation mosaicism in males with Fragile X syndrome. Expert Rev. Mol. Diagn. 2017, 17, 1023–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milà, M.; Castellví-Bel, S.; Sánchez, A.; Lázaro, C.; Villa, M.; Estivill, X. Mosaicism for the fragile X syndrome full mutation and deletions within the CGG repeat of the FMR1 gene. J. Med. Genet. 1996, 33, 338–340. [Google Scholar] [CrossRef] [Green Version]
- Grasso, M.; Faravelli, F.; Lo Nigro, C.; Chiurazzi, P.; Sperandeo, M.P.; Argusti, A.; Pomponi, M.G.; Lecora, M.; Sebastio, G.F.; Perroni, L.; et al. Mosaicism for the full mutation and a microdeletion involving the CGG repeat and flanking sequences in the FMR1 gene in eight fragile X patients. Am. J. Med. Genet. 1999, 85, 311–316. [Google Scholar] [CrossRef]
- Hwang, Y.T.; Aliaga, S.M.; Arpone, M.; Francis, D.; Li, X.; Chong, B.; Slater, H.R.; Rogers, C.; Bretherton, L.; Hunter, M.; et al. Partially methylated alleles, microdeletion, and tissue mosaicism in a fragile X male with tremor and ataxia at 30 years of age: A case report. Am. J. Med. Genet. A 2016, 170, 3327–3333.32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petek, E.; Kroisel, P.M.; Schuster, M.; Zierler, H.; Wagner, K. Mosaicism in a fragile X male including a de novo deletion in the FMR1 gene. Am. J. Med. Genet. 1999, 84, 229–232. [Google Scholar] [CrossRef]
- Fan, H.; Booker, J.K.; McCandless, S.E.; Shashi, V.; Fleming, A.; Farber, R.A. Mosaicism for an FMR1 gene deletion in a fragile X female. Am. J. Med. Genet. A 2005, 136, 214–217. [Google Scholar] [CrossRef]
- Govaerts, L.C.; Smit, A.E.; Saris, J.J.; VanderWerf, F.; Willemsen, R.; Bakker, C.E.; De Zeeuw, C.I.; Oostra, B.A. Exceptional good cognitive and phenotypic profile in a male carrying a mosaic mutation in the FMR1 gene. Clin. Genet. 2007, 72, 138–144. [Google Scholar] [CrossRef] [PubMed]
- De Vries, B.B.; Fryns, J.P.; Butler, M.G.; Canziani, F.; Wesby-van Swaay, E.; van Hemel, J.O.; Oostra, B.A.; Halley, D.J.; Niermeijer, M.F. Clinical and molecular studies in fragile X patients with a Prader-Willi-like phenotype. J. Med. Genet. 1993, 30, 761–766. [Google Scholar] [CrossRef] [PubMed]
- De Vries, B.B.; Wiegers, A.M.; de Graaff, E.; Verkerk, A.J.; Van Hemel, J.O.; Halley, D.J.; Fryns, J.P.; Curfs, L.M.; Niermeijer, M.F.; Oostra, B.A. Mental status and fragile X expression in relation to FMR-1 gene mutation. Eur. J. Hum. Genet. 1993, 1, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Han, X.D.; Powell, B.R.; Phalin, J.L.; Chehab, F.F. Mosaicism for a full mutation, premutation, and deletion of the CGG repeats results in 22% FMRP and elevated FMR1 mRNA levels in a high-functioning fragile X male. Am. J. Med. Genet. A 2006, 140, 1463–1471. [Google Scholar] [CrossRef] [PubMed]
- Mannermaa, A.; Pulkkinen, L.; Kajanoja, E.; Ryynänen, M.; Saarikoski, S. Deletion in the FMR1 gene in a fragile-X male. Am. J. Med. Genet. 1996, 64, 293–295. [Google Scholar] [CrossRef]
- De Graaff, E.; de Vries, B.B.; Willemsen, R.; van Hemel, J.O.; Mohkamsing, S.; Oostra, B.A.; van den Ouweland, A.M. The fragile X phenotype in a mosaic male with a deletion showing expression of the FMR1 protein in 28% of the cells. Am. J. Med. Genet. 1996, 64, 302–308. [Google Scholar] [CrossRef]
- Perroni, L.; Grasso, M.; Argusti, A.; Lo Nigro, C.; Croci, G.F.; Zelante, L.; Garani, G.P.; Dagna Bricarelli, F. Molecular and cytogenetic analysis of the fragile X syndrome in a series of 453 mentally retarded subjects: A study of 87 families. Am. J. Med. Genet. 1996, 64, 176–180. [Google Scholar] [CrossRef]
- Park, K.M.; Jun, K.R.; Lee, H.J.; Park, S.; Kim, S.E. Unilateral diffuse white matter involvement in a patient with atypical FMR1 mutation. Clin. Neurol. Neurosurg. 2020, 197, 106182. [Google Scholar] [CrossRef]
- Grønskov, K.; Brøndum-Nielsen, K.; Dedic, A.; Hjalgrim, H. A nonsense mutation in FMR1 causing fragile X syndrome. Eur. J. Hum. Genet. 2011, 19, 489–491. [Google Scholar] [CrossRef]
- Maddirevula, S.; Alsaif, H.S.; Ibrahim, N.; Alkuraya, F.S. A de novo mutation in FMR1 in a patient with intellectual disability. Eur. J. Med. Genet. 2020, 63, 103763. [Google Scholar] [CrossRef]
- Saldarriaga, W.; Payán-Gómez, C.; González-Teshima, L.Y.; Rosa, L.; Tassone, F.; Hagerman, R.J. Double Genetic Hit: Fragile X Syndrome and Partial Deletion of Protein Patched Homolog 1 Antisense as Cause of Severe Autism Spectrum Disorder. J. Dev. Behav. Pediatr. 2020, 41, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Lugenbeel, K.A.; Peier, A.M.; Carson, N.L.; Chudley, A.E.; Nelson, D.L. Intragenic loss of function mutations demonstrate the primary role of FMR1 in fragile X syndrome. Nat. Genet. 1995, 10, 483–485. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.F.; Fitzgerald, T.W.; Jones, W.D.; Clayton, S.; McRae, J.F.; van Kogelenberg, M.; King, D.A.; Ambridge, K.; Barrett, D.M.; Bayzetinova, T.; et al. Genetic diagnosis of developmental disorders in the DDD study: A scalable analysis of genome-wide research data. Lancet 2015, 385, 1305–1314. [Google Scholar] [CrossRef] [Green Version]
- Collins, S.C.; Bray, S.M.; Suhl, J.A.; Cutler, D.J.; Coffee, B.; Zwick, M.E.; Warren, S.T. Identification of novel FMR1 variants by massively parallel sequencing in developmentally delayed males. Am. J. Med. Genet. A 2010, 152a, 2512–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myrick, L.K.; Deng, P.Y.; Hashimoto, H.; Oh, Y.M.; Cho, Y.; Poidevin, M.J.; Suhl, J.A.; Visootsak, J.; Cavalli, V.; Jin, P.; et al. Independent role for presynaptic FMRP revealed by an FMR1 missense mutation associated with intellectual disability and seizures. Proc. Natl. Acad. Sci. USA 2015, 112, 949–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitzmann, A.F.; Hagelstrom, R.T.; Tassone, F.; Hagerman, R.J.; Butler, M.G. Rare FMR1 gene mutations causing fragile X syndrome: A review. Am. J. Med. Genet. A 2018, 176, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Prieto, M.; Folci, A.; Poupon, G.; Schiavi, S.; Buzzelli, V.; Pronot, M.; François, U.; Pousinha, P.; Lattuada, N.; Abelanet, S.; et al. Missense mutation of Fmr1 results in impaired AMPAR-mediated plasticity and socio-cognitive deficits in mice. Nat. Commun. 2021, 12, 1557. [Google Scholar] [CrossRef]
- Diaz, J.; Scheiner, C.; Leon, E. Presentation of a recurrent FMR1 missense mutation (R138Q) in an affected female. Transl. Sci. Rare Dis. 2018, 3, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Myrick, L.K.; Nakamoto-Kinoshita, M.; Lindor, N.M.; Kirmani, S.; Cheng, X.; Warren, S.T. Fragile X syndrome due to a missense mutation. Eur. J. Hum. Genet. 2014, 22, 1185–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boulle, K.; Verkerk, A.J.; Reyniers, E.; Vits, L.; Hendrickx, J.; Van Roy, B.; Van den Bos, F.; de Graaff, E.; Oostra, B.A.; Willems, P.J. A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat. Genet. 1993, 3, 31–35. [Google Scholar] [CrossRef]
- Feng, Y.; Absher, D.; Eberhart, D.E.; Brown, V.; Malter, H.E.; Warren, S.T. FMRP associates with polyribosomes as an mRNP, and the I304N mutation of severe fragile X syndrome abolishes this association. Mol. Cell 1997, 1, 109–118. [Google Scholar] [CrossRef]
- Carroll, R.; Shaw, M.; Arvio, M.; Gardner, A.; Kumar, R.; Hodgson, B.; Heron, S.; McKenzie, F.; Järvelä, I.; Gecz, J. Two novel intragenic variants in the FMR1 gene in patients with suspect clinical diagnosis of Fragile X syndrome and no CGG repeat expansion. Eur. J. Med. Genet. 2020, 63, 104010. [Google Scholar] [CrossRef]
- Zeidler, S.; Severijnen, L.A.; de Boer, H.; van der Toorn, E.C.; Ruivenkamp, C.A.L.; Bijlsma, E.K.; Willemsen, R. A missense variant in the nuclear export signal of the FMR1 gene causes intellectual disability. Gene 2021, 768, 145298. [Google Scholar] [CrossRef] [PubMed]
- Handt, M.; Epplen, A.; Hoffjan, S.; Mese, K.; Epplen, J.T.; Dekomien, G. Point mutation frequency in the FMR1 gene as revealed by fragile X syndrome screening. Mol. Cell Probes 2014, 28, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Okray, Z.; de Esch, C.E.; Van Esch, H.; Devriendt, K.; Claeys, A.; Yan, J.; Verbeeck, J.; Froyen, G.; Willemsen, R.; de Vrij, F.M.; et al. A novel fragile X syndrome mutation reveals a conserved role for the carboxy-terminus in FMRP localization and function. EMBO Mol. Med. 2015, 7, 423–437. [Google Scholar] [CrossRef]
- Wang, Y.C.; Lin, M.L.; Lin, S.J.; Li, Y.C.; Li, S.Y. Novel point mutation within intron 10 of FMR-1 gene causing fragile X syndrome. Hum. Mutat. 1997, 10, 393–399. [Google Scholar] [CrossRef]
- Tarleton, J.; Kenneson, A.; Taylor, A.K.; Crandall, K.; Fletcher, R.; Casey, R.; Hart, P.S.; Hatton, D.; Fisch, G.; Warren, S.T. A single base alteration in the CGG repeat region of FMR1: Possible effects on gene expression and phenotype. J. Med. Genet. 2002, 39, 196–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grønskov, K.; Hallberg, A.; Brøndum-Nielsen, K. Mutational analysis of the FMR1 gene in 118 mentally retarded males suspected of fragile X syndrome: Absence of prevalent mutations. Hum. Genet. 1998, 102, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Golden, C.E.M.; Breen, M.S.; Koro, L.; Sonar, S.; Niblo, K.; Browne, A.; Burlant, N.; Di Marino, D.; De Rubeis, S.; Baxter, M.G.; et al. Deletion of the KH1 Domain of Fmr1 Leads to Transcriptional Alterations and Attentional Deficits in Rats. Cereb. Cortex 2019, 29, 2228–2244. [Google Scholar] [CrossRef] [Green Version]
- Carion, N.; Briand, A.; Cuisset, L.; Pacot, L.; Afenjar, A.; Bienvenu, T. Loss of the KH1 domain of FMR1 in humans due to a synonymous variant causes global developmental retardation. Gene 2020, 753, 144793. [Google Scholar] [CrossRef] [PubMed]
- Shinahara, K.; Saijo, T.; Mori, K.; Kuroda, Y. Single-strand conformation polymorphism analysis of the FMR1 gene in autistic and mentally retarded children in Japan. J. Med. Investig. 2004, 51, 52–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, J.B.; Gurling, H.M. Point mutation in intron 10 of FMR1 is unlikely to be a cause of fragile X syndrome. Hum. Mutat. 1998, 12, 431–432. [Google Scholar] [CrossRef]
- Vincent, J.B.; Konecki, D.S.; Munstermann, E.; Bolton, P.; Poustka, A.; Poustka, F.; Gurling, H.M. Point mutation analysis of the FMR-1 gene in autism. Mol. Psychiatry 1996, 1, 227–231. [Google Scholar] [PubMed]
- Vincent, J.B.; Thevarkunnel, S.; Kolozsvari, D.; Paterson, A.D.; Roberts, W.; Scherer, S.W. Association and transmission analysis of the FMR1 IVS10 + 14C-T variant in autism. Am. J. Med. Genet. B Neuropsychiatr Genet. 2004, 125B, 54–56. [Google Scholar] [CrossRef]
- Suhl, J.A.; Muddashetty, R.S.; Anderson, B.R.; Ifrim, M.F.; Visootsak, J.; Bassell, G.J.; Warren, S.T. A 3’ untranslated region variant in FMR1 eliminates neuronal activity-dependent translation of FMRP by disrupting binding of the RNA-binding protein HuR. Proc. Natl. Acad. Sci. USA 2015, 112, E6553–E6561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.G.; Yan, A.Z.; Xu, Y.J.; Liao, J.; Guo, X.Y.; Zhang, D.; Yang, W.J.; Zheng, D.Z.; Lan, F.H. Splicing of exon 9a in FMR1 transcripts results in a truncated FMRP with altered subcellular distribution. Gene 2020, 731, 144359. [Google Scholar] [CrossRef]
- Bardoni, B.; Sittler, A.; Shen, Y.; Mandel, J.L. Analysis of domains affecting intracellular localization of the FMRP protein. Neurobiol. Dis. 1997, 4, 329–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, E.; Rajan, N.; Bagni, C. The FMRP regulon: From targets to disease convergence. Front. Neurosci. 2013, 7, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myrick, L.K.; Hashimoto, H.; Cheng, X.; Warren, S.T. Human FMRP contains an integral tandem Agenet (Tudor) and KH motif in the amino terminal domain. Hum. Mol. Genet. 2015, 24, 1733–1740. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, S.; Ramos, A.; Martin, S.R.; Dal Piaz, F.; Pucci, P.; Bardoni, B.; Mandel, J.L.; Pastore, A. The N-terminus of the fragile X mental retardation protein contains a novel domain involved in dimerization and RNA binding. Biochemistry 2003, 42, 10437–10444. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, M.; Nalavadi, V.; Epstein, M.P.; Narayanan, U.; Bassell, G.J.; Warren, S.T. Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 15537–15542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff, D.J.; Gustashaw, K.M.; Zurcher, V.; Ko, L.; White, W.; Weiss, L.; Van Dyke, D.L.; Schwartz, S.; Willard, H.F. Deletions in Xq26.3-q27.3 including FMR1 result in a severe phenotype in a male and variable phenotypes in females depending upon the X inactivation pattern. Hum. Genet. 1997, 100, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Zink, A.M.; Wohlleber, E.; Engels, H.; Rødningen, O.K.; Ravn, K.; Heilmann, S.; Rehnitz, J.; Katzorke, N.; Kraus, C.; Blichfeldt, S.; et al. Microdeletions including FMR1 in three female patients with intellectual disability—Further delineation of the phenotype and expression studies. Mol. Syndromol. 2014, 5, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Aiba, K.; Fukushi, D.; Yoshimura, J.; Suzuki, Y.; Mitsui, J.; Morishita, S.; Tuji, S.; Yamada, K.; Wakamatsu, N. Clinical and molecular genetic characterization of two female patients harboring the Xq27.3q28 deletion with different ratios of X chromosome inactivation. Hum. Mutat. 2020, 41, 1447–1460. [Google Scholar] [CrossRef]
- Schmidt, M.; Certoma, A.; Du Sart, D.; Kalitsis, P.; Leversha, M.; Fowler, K.; Sheffield, L.; Jack, I.; Danks, D.M. Unusual X chromosome inactivation in a mentally retarded girl with an interstitial deletion Xq27: Implications for the fragile X syndrome. Hum. Genet. 1990, 84, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Birot, A.M.; Delobel, B.; Gronnier, P.; Bonnet, V.; Maire, I.; Bozon, D. A 5-megabase familial deletion removes the IDS and FMR-1 genes in a male Hunter patient. Hum. Mutat. 1996, 7, 266–268. [Google Scholar] [CrossRef]
- Probst, F.J.; Roeder, E.R.; Enciso, V.B.; Ou, Z.; Cooper, M.L.; Eng, P.; Li, J.; Gu, Y.; Stratton, R.F.; Chinault, A.C.; et al. Chromosomal microarray analysis (CMA) detects a large X chromosome deletion including FMR1, FMR2, and IDS in a female patient with mental retardation. Am. J. Med. Genet. A 2007, 143a, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Burruss, D.M.; Wood, T.C.; Espinoza, L.; Dwivedi, A.; Holden, K.R. Severe Hunter syndrome (mucopolysaccharidosis II) phenotype secondary to large deletion in the X chromosome encompassing IDS, FMR1, and AFF2 (FMR2). J. Child. Neurol. 2012, 27, 786–790. [Google Scholar] [CrossRef] [PubMed]
- Marshall, L.S.; Simon, J.; Wood, T.; Peng, M.; Owen, R.; Feldman, G.S.; Zaragoza, M.V. Deletion Xq27.3q28 in female patient with global developmental delays and skewed X-inactivation. BMC Med. Genet. 2013, 14, 49. [Google Scholar] [CrossRef] [Green Version]
- Stoof, S.C.M.; Kersseboom, R.; de Vries, F.A.T.; Kruip, M.; Kievit, A.J.A.; Leebeek, F.W.G. Hemophilia B in a female with intellectual disability caused by a deletion of Xq26.3q28 encompassing the F9. Mol. Genet. Genom. Med. 2018, 6, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Kitada, K.; Santostefano, K.E.; Yokoyama, A.; Waldrop, S.M.; Heldermon, C.D.; Tachibana, D.; Koyama, M.; Meacham, A.M.; Pacak, C.A.; et al. Generation of Induced Pluripotent Stem Cells from a Female Patient with a Xq27.3-q28 Deletion to Establish Disease Models and Identify Therapies. Cell Reprogram. 2020, 22, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Yachelevich, N.; Gittler, J.K.; Klugman, S.; Feldman, B.; Martin, J.; Brooks, S.S.; Dobkin, C.; Nolin, S.L. Terminal deletions of the long arm of chromosome X that include the FMR1 gene in female patients: A case series. Am. J. Med. Genet. A 2011, 155a, 870–874. [Google Scholar] [CrossRef]
- Hickey, S.E.; Walters-Sen, L.; Mosher, T.M.; Pfau, R.B.; Pyatt, R.; Snyder, P.J.; Sotos, J.F.; Prior, T.W. Duplication of the Xq27.3-q28 region, including the FMR1 gene, in an X-linked hypogonadism, gynecomastia, intellectual disability, short stature, and obesity syndrome. Am. J. Med. Genet. A 2013, 161a, 2294–2299. [Google Scholar] [CrossRef] [PubMed]
- Rio, M.; Malan, V.; Boissel, S.; Toutain, A.; Royer, G.; Gobin, S.; Morichon-Delvallez, N.; Turleau, C.; Bonnefont, J.P.; Munnich, A.; et al. Familial interstitial Xq27.3q28 duplication encompassing the FMR1 gene but not the MECP2 gene causes a new syndromic mental retardation condition. Eur. J. Hum. Genet. 2010, 18, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Erster, S.H.; Brown, W.T.; Goonewardena, P.; Dobkin, C.S.; Jenkins, E.C.; Pergolizzi, R.G. Polymerase chain reaction analysis of fragile X mutations. Hum. Genet. 1992, 90, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.T.; Houck, G.E., Jr.; Jeziorowska, A.; Levinson, F.N.; Ding, X.; Dobkin, C.; Zhong, N.; Henderson, J.; Brooks, S.S.; Jenkins, E.C. Rapid fragile X carrier screening and prenatal diagnosis using a nonradioactive PCR test. JAMA 1993, 270, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Pieretti, M.; Sutcliffe, J.S.; Richards, S.; Verkerk, A.J.; Holden, J.J.; Fenwick, R.G., Jr.; Warren, S.T.; et al. Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell 1991, 67, 1047–1058. [Google Scholar] [CrossRef]
- Kremer, E.J.; Pritchard, M.; Lynch, M.; Yu, S.; Holman, K.; Baker, E.; Warren, S.T.; Schlessinger, D.; Sutherland, G.R.; Richards, R.I. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science 1991, 252, 1711–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Mulley, J.; Loesch, D.; Turner, G.; Donnelly, A.; Gedeon, A.; Hillen, D.; Kremer, E.; Lynch, M.; Pritchard, M.; et al. Fragile-X syndrome: Unique genetics of the heritable unstable element. Am. J. Hum. Genet. 1992, 50, 968–980. [Google Scholar] [PubMed]
- Hegde, M.R.; Chong, B.; Fawkner, M.; Lambiris, N.; Peters, H.; Kenneson, A.; Warren, S.T.; Love, D.R.; McGaughran, J. Microdeletion in the FMR-1 gene: An apparent null allele using routine clinical PCR amplification. J. Med. Genet. 2001, 38, 624–629. [Google Scholar] [CrossRef] [Green Version]
- Stosser, M.B.; Lindy, A.S.; Butler, E.; Retterer, K.; Piccirillo-Stosser, C.M.; Richard, G.; McKnight, D.A. High frequency of mosaic pathogenic variants in genes causing epilepsy-related neurodevelopmental disorders. Genet. Med. 2018, 20, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Tejada, M.I.; Glover, G.; Martínez, F.; Guitart, M.; de Diego-Otero, Y.; Fernández-Carvajal, I.; Ramos, F.J.; Hernández-Chico, C.; Pintado, E.; Rosell, J.; et al. Molecular testing for fragile X: Analysis of 5062 tests from 1105 fragile X families--performed in 12 clinical laboratories in Spain. BioMed Res. Int. 2014, 2014, 195793. [Google Scholar] [CrossRef] [PubMed]
- Mantere, T.; Kersten, S.; Hoischen, A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019, 10, 426. [Google Scholar] [CrossRef] [Green Version]
- Ho, S.S.; Urban, A.E.; Mills, R.E. Structural variation in the sequencing era. Nat. Rev. Genet. 2020, 21, 171–189. [Google Scholar] [CrossRef] [PubMed]
- Dolzhenko, E.; Bennett, M.F.; Richmond, P.A.; Trost, B.; Chen, S.; van Vugt, J.; Nguyen, C.; Narzisi, G.; Gainullin, V.G.; Gross, A.M.; et al. ExpansionHunter Denovo: A computational method for locating known and novel repeat expansions in short-read sequencing data. Genome Biol. 2020, 21, 102. [Google Scholar] [CrossRef] [PubMed]
- Dolzhenko, E.; Deshpande, V.; Schlesinger, F.; Krusche, P.; Petrovski, R.; Chen, S.; Emig-Agius, D.; Gross, A.; Narzisi, G.; Bowman, B.; et al. ExpansionHunter: A sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics 2019, 35, 4754–4756. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Ref. | Location b | Number Affected with Variant, Sex (M/F) | Inheritance/Originating Allele | Ascertainment | ACMG Criteria [12,13] | Conclusion | ||
|---|---|---|---|---|---|---|---|---|
| Proximal | Distal | Type | ||||||
| [15] | >140198205 (DXS1232-DXS105), <7 Mb upstream | <148215436 (between 141R-DXS533), 264 kb downstream | Del | 1M | Maternal (unaffected), de novo in mother (both grandparents lack deletion with normal CGG repeat counts) | ID, obesity | CNV 2A 5G_0.1 | PATH (1.1) |
| [16] | >140784366 (CDR1-sWXD2905), <7 Mb upstream | <148203554 (DXS7847); >DXS8318, <252 kb downstream | Del | 1M + mother | Maternal (mildly affected) | DD, clinical suspicion | CNV 2A 5G_0.1 | PATH (1.1) |
| [17] #3 | 142254456, 5.7 Mb upstream | 148191426, 240 kb downstream | Del het | 1F | De novo | Epilepsy research database | CNV 2A 5A_0.15 | PATH (1.15) |
| [18] | 147158490, 753 kb upstream | 148171882, 221 kb downstream | Mosaic del (90%) | 1M | Mosaic in proband on 23-repeat allele (mother 23, 30 repeats) with breakpoints in LINE1 elements | ID | CNV 2A 5A_0.15 | PATH (1.15) |
| [19] | 147630212–147721930 (DXS532–DXS548), 200–300 kb upstream | Intragenic, ~30 kb downstream of HTF island | Del | 1M | De novo | ID, facies, testicle size | SVI PVS1_Strong PS2 PM2 | PATH |
| [20] | 147653688 c, 260 kb upstream | 147955394, 4 kb downstream | Del | 1M | Mosaic mother (3/400 lymphocytes) with same deletion in subsequent pregnancy | Clinical FXS | CNV 2A 5H_0.1 | PATH (1.1) |
| [21] | >147722126 (DXS548); >DXS477, <cosmid 494; <190 kb upstream | 147932685–147932763, exon 9 | Del | 1M | ? (mother “borderline intelligence”) | ID | SVI PVS1_Strong PM2 | LPATH |
| [22] #2 | >147722126 (DXS548); >G9L, <FRAXAC1; <190 kb upstream | 147936614–147937465, intron 10 | Del | 1M | De novo on 19-repeat allele (mother 19, 51 repeats) | ID, clinical FXS | SVI PVS1_Strong PS2 PS3 PM2 | PATH |
| [23] | <147722126 (includes DXS548), not mapped further; >190 kb upstream | ?, includes FMR1 | Del | 1M + infant brother | Mat germline mosaicism (2 brothers, absent in mother) (mother has congenital digit amputations) | DD | CNV 2A 5G_0.1 | PATH (1.1) |
| [24] #18072 | 147787231, 125 kb upstream | 148041310, 90 kb downstream | Del | 2M | Maternal (unaffected het) | Autism cohort | CNV 2A5G_0.1 | PATH (1.1) |
| [25] #3 | 147838064, 74 kb upstream | 148103912, 400 kb downstream | Del | 1M | De novo | Clinical lab sample | CNV 2A 5A_0.15 | PATH (1.15) |
| [26] | 15–80 kb upstream (G9L YAC) | 147930245–147932424, intron 7 | Del | 1M | De novo (normal repeat alleles in mother) | DD | SVI PVS1_Strong PS2 PS3 PM2 | PATH |
| [27,28] | 4.4 kb upstream | 194 kb downstream | Del | 1M | Maternal (unaffected mosaic) d; breakpoints in L1MC2 and MIR3 elements | Preschool cohort with ID and >=1 FXS feature | CNV 2A 5G_0.1 | PATH (1.1) |
| [29] #24 | 147910365, 1.5 kb upstream | 147912050 (within CGG repeat, no AGG interspersions in remaining sequence) | Del | 4M, 2F | Maternal (unaffected) | Speech delay, hyperactivity | CNV 2C2_0.3 5D_0.3 | VUS (0.6) |
| [22] #1 | 147911457, 462 bp upstream of transcription start | 147912135, c.-45 (including entire CGG repeat) | Mosaic del (40%) | 1M | De novo | Epilepsy, other clinical | CNV 2C2_0.3 5A_0.15 | VUS (0.45) |
| [30] | ~147911751 (~300 bp upstream of CGG) | ? (~400 bp size deletion) | Del | 1M | De novo (mother is het for 700–900 repeat full mutation) | ID, aggressive behavior | CNV 2C2_0 5A_0.15 | VUS (0.15) |
| Also has 13p+ polymorphism | ||||||||
| [31] | 147911831, 88 bp upstream of transcription start | 147912185, c.6 | Del | 1M | ID male with >=2 FXS features | SVI PVS1_Moderate PS3 PM2 | LPATH | |
| [32] | 147911966, c.-214 | Within CGG repeats, 19 remaining (no AGGs) | Del | N/A (M) | Maternal mosaic (unaffected het for 430–530 repeat full mutation, deletion occurred on full mutation allele by haplotype) | Unaffected, prenatal testing | CNV 2C2_0 5F_0 | VUS (0) |
| [33] | 147911981, c.-199 | 147912050 (within CGG repeats, 9 remaining) | Del het | N/A | Transmitted from unaffected female proband to male fetus | Unaffected individual, population screening study | CNV 2C2_0 5F_0 | VUS (0) |
| [34] | 147911984, c.-196 | 147912140, c.-40 (distal to CGG repeat) | Del het | N/A (F) | De novo, from maternal full mutation allele | Asymptomatic; due to brother with FXS | CNV 2C2_0 5A_-0.3; lymphoblasts are FMRP+ | VUS (−0.3) with additional benign evidence |
| In trans with large deletion involving FMR1-46,X,del(X)(q24) | ||||||||
| [35] | 147912024, c.-156 | 147912111, c.-69 (1 bp distal to CGG repeat) | Del | N/A (M) | De novo, from maternal full mutation allele | Asymptomatic; due to maternal full mutation | CNV 2C2_0 5A_-0.3 (unaffected); normal blood FMR1 mRNA level | VUS (−0.3) with additional benign evidence |
| [36] APN26, [37] #1 | <147948682 (Deletion of exon 17) | >147964837 (Deletion of exon 17) | Del | 3M | Maternal (unaffected) | ID sequencing cohort | SVI PVS1_Moderate PS3_Supporting PM2 PP1 | LPATH |
| [33] | Undetermined 5′ UTR | Loss of exon 1 | Del | 1M fetus | Mother is full mutation (280 repeat) het | Population screening study | SVI PVS1_Strong PS3 | PATH |
| [38] #3660 | Intragenic (~197 bp flanking CGG repeat in exon 1) | Intragenic | Del | 1M | ? (mother has mild ID) | ID cohort | CNV 2C2_0 5G_0.1 | VUS (0.1) |
| Potentially including AFF2 (additional reports of confirmed FMR1-AFF2 deletions not shown) | ||||||||
| [25] #4 | 142773651, 5.1 Mb upstream | 148673162, 722 kb downstream | Del het | 1F | De novo | Clinical lab sample | CNV 2A 5A_0.15 | PATH (1.15) |
| [17] #2 | 143096757, 4.8 Mb upstream | 149186971, 1.2 Mb downstream | Del het | 1F | De novo | Epilepsy research database | CNV 2A 5A_0.15 | PATH (1.15) |
| Duplications | ||||||||
| [25] #1 | 147707513 (204 kb upstream) | 148070742 (120 kb downstream) | Dup het | 1 | De novo | Clinical testing (lab) | CNV 2H_0 4C_0.15x3 5A_0.15 | VUS (0.6) |
| [25] | 147709189 (203 kb upstream) | 147976294 (25 kb downstream) | Dup | 1 | Clinical testing for seizures (lab) | CNV 2H_0 4C_0.15x3 5A_0.15 | VUS (0.6) | |
| [25] #2 | 147796927 (115 kb upstream) | 148144967 (194 kb downstream) | Dup het | 1 | De novo | Clinical testing (lab) | CNV 2H_0 4C_0.15x3 5A_0.15 | VUS (0.6) |
| [39] | 147894723 (17 kb upstream) | 147979356 (28 kb downstream) | Dup | 1 | De novo (Proband also has small 1q44 paternal dup and 4p15 del) | Myoclonic seizures | CNV 2H_0 4C_0.15x3 5A_0.15; normal mRNA in leukocytes | VUS (0.6) |
| [40] | 147912003 (c.-177) | 147912051 (c.-129) | Dup | N/A (0.2–0.3% X chromosomes in study) | Finnish population samples with multiple unaffected males | CNV 2I_0 4O_-1 | BEN (−1) | |
| Ref./ Case # | Breakpoints a | c. | Sex | Expansion | % Deletion in Blood | Functional/ Phenotypes b | Maternal CGG Repeats | ||
|---|---|---|---|---|---|---|---|---|---|
| Proximal | Distal | Proximal | Distal | ||||||
| [48] | 147911612 | 147912529 | upstream | c.51+299 | M | 300–350 | No FMRP (blood, hair roots) | ~100, normal | |
| [45] #1513 | ~147911695 | ~147912736 | upstream | ~c.51+506 | M | FM | 3–4% mRNA | 99/105, 30 | |
| [49] | 147911883 | 147912368 | upstream | c.51+138 | M | FM | 20–30% | ||
| [50] #12 | 147911908(ins(9)) | 147912135 | upstream | c.-45 | M | 250, 510, 710; 170–320 with deletion | 9% (with 22.5% unmethylated, 68.5% methylated) | 11% FMRP, 72% mRNA; Mullen early learning composite 90 | |
| [51] #1 | ~147911938 | CGG repeat | ~c.-242 | 30 repeats | M | 300–500 | 17% | 156, 23 | |
| [50] #11 | 147911941 | 147912291 | c.-239 | c.51+61 | M | 420, 470, 1040; 84 with deletion | 20% (with 13% unmethylated, 67% methylated) | 6% FMRP, 17% mRNA; IQ 66 | |
| [52] #6 | ~147911945 | 147912267 | ~c.-235 | c.51+37 | M | FM | No FMRP (lymphocytes) | FM, normal | |
| [50] #9 | 147911960 (insTT) | 147912133 | c.-220 | c.-47 | M | 350, 510, 630, 1050; 150–500 with deletion | 13% (with 28% unmethylated, 59% methylated) | 32% FMRP, 64% mRNA; IQ 47, autism spectrum disorder | |
| [53] | ~147911964 | ~147912164 | ~c.-216 | ~c.-16 | M | 70, 463–846 | FXS + FXTAS 97% FMRP (blood) | PM/FM mosaic, normal | |
| [52] #5 | ~147911966 | 147912125 ins(7) | ~c.-214 | c.-55 | M | FM | No FMRP (lymphocytes) | FM, normal | |
| [46] #2 | 147911966 | 147912135 | c.-214 | c.-45 | M | FM | 5–10% | PM, normal | |
| [47] | 147911966 | 147920712 | c.-214 | c.51+8482 | M | ~300 | 90%; 85% fibroblasts (12/14) | FM, normal (affected) | |
| [54] | 147911976 | 147912202 | c.-204 | c.23 | M | 58–60, FM | 2% (23% PM, 75% FM) | ||
| [46] #4 | 147911978 | 147912134 | c.-202 | c.-46 | M | FM | 5–10% | ||
| [46] #3 | 147911978 | ~147912545 | c.-202 | ~c.51+315 | M | FM | 15% | ||
| [55] | 147911988 | 147912198 | c.-192 | c.19 | F | ~400/normal het | 21% (29% FM, 27% methylated normal, 23% unmethylated normal) | PM, normal | |
| [50] #10 | 147911990 (insCG) | 147912138 | c.-190 | c.-42 | M | 450, 540, 620, 930, 1080; 118 with deletion | 4% (with 14% unmethylated, 82% methylated) | 5% FMRP, 14% mRNA; IQ 69 | |
| [56] | ~147911990 | ~147912137 | ~c.-190 | ~ c.-97 | M | 170, 230, 450 | 15% (60% methylated FM, 25% unmethylated PM) | 18% FMRP (lymphocytes), 6% FMRP (hair roots), mild (IQ 81) | PM, normal |
| [46] #1, [57] #2, [58] | 147911998 | 147912288 | c.-182 | c.51+58 | M | FM | 5–10% | Prader-Willi syndrome phenotype | PM, normal |
| [59] | 147912010 | 147912111 | c.-170 | c.-69 | M | 165, >200 | 53% (Southern; with 37% PM, 10% FM) | 22% WBCs are FMRP+ Milder FXS than non-mosaic brother | 109, normal |
| [60] | 147912020 | 147912171 | c.-160 | c.-9 | M | FM | |||
| [45] #1629 | 147912023 | 147912115 | c.-157 | c.-65 | M | FM | PM, normal | ||
| [45] #1033 | 147912023 | 147912150 | c.-157 | c.-30 | M | FM | 3% mRNA | 99/106, 31 | |
| [45] #1337 | 147912039 | 147912110 insCTGGG | c.-141 | c.-70 | M | FM | 16–19% mRNA | 79, 30 | |
| [45] #1234 | 147912044 | 147912136 | c.-136 | c.-44 | M | FM | 4–6% mRNA | 174/>200, 36 | |
| [61] | ~147912060 | 147912140 | ~c.-120 (4th CGG) | c.-40 | M | FM | 20–30% | 28% lymphocytes are FMRP+ Typical FXS | |
| [52] #7 | >147909424 (RFLP) | <147912612 (RFLP) | upstream | <c.51+382 | M | FM | No FMRP (lymphoblastoid cell line) | PM, normal | |
| [62] | >147911959 (primer) | <147912181 (primer) | >c.-221 | <c.2 | M | FM | |||
| [52] #8 | ? (RFLP) | ? (RFLP) | entire repeat | M | FM | FM, normal | |||
| Ref. | Location | Number Affected with Variant, Sex (M/F) | Other Variants in Patient | Inheritance in Proband | ACMG Criteria [11] | Conclusion | |
|---|---|---|---|---|---|---|---|
| c. | p. | ||||||
| [64] | c.80C>A | p.S27* | 1M, 1F | Mat (affected) | PVS1 PS3 PM2 PP1 | PATH | |
| [65] | c.188_193del | p.(D63_E64del) | 1M | De novo confirmed | PS2 PM2 PM4 PP3 | LPATH | |
| [66] | c.229delT | p.(C77Afs*5) | 1M | Maternal PATH 308 kb del involving PTCHD1-AS (autism) | De novo confirmed | PVS1 PS2 PM2 BP5 | PATH |
| [67] #1 | c.373delA a | p.(T125Lfs*35) | 1M | De novo | PVS1 PS3 PM6 | PATH | |
| [37,68] b | c.377T>C | p.(F126S) | 1M | KDM5C I1349M (maternal), ALG13 H771R (maternal) | De novo | PS2 PM6 PP3 | LPATH |
| [69,70] | c.413G>A | p.R138Q | 1M, 1F | Maternal (affected, learning disability/anxiety) | PS3 PP1 PP4; note 7 hemizygotes in gnomAD v3 exomes (rs200163413) | LPATH | |
| [71] | c.413G>A | p.R138Q | 1M | (1st cousin consanguinity) | Maternal | ||
| [72] | c.413G>A | p.R138Q | N/A | ||||
| [73] | c.413G>A | p.R138Q | 1F | Absent in mother; healthy father not tested | |||
| [74] | c.797G>A | p.(G266E) | 1M | Maternal (unaffected) | PS3 PM2 PP3 | LPATH | |
| [75,76] | c.911T>A | p.(I304N) | 1M | Known PHKA2 deficiency (glycogen storage disease IXa1) | De novo confirmed | PS2 PS3 PM2 PP3 | PATH |
| [77] #1 | c.1021–1028delinsTATTGG | p.N341Yfs*7 | 2M | Mother not tested; had epilepsy | PVS1 PS3 PP1 | PATH | |
| [78] | c.1325G>A | p.(R442Q) | 2M + mother, grandmother | 11 kb paternal deletion of part of MYH7 | Maternal (affected) | PS3 PM2 PP1 PP3 | LPATH |
| [79] | c.1444G>A | p.(G482S) | 1M | PM2 BP4 | VUS | ||
| [63] | c.1550C>T c | p.(P517L) | 1M | ? | PM2 | VUS | |
| [79] | c.1601G>A | p.(R534H) | 2M (unrelated), 1F | Maternal (unaffected) in both families | PM2 | VUS | |
| [80] | c.1610dup | p.(G538Rfs*24) | 1M | ? | PVS1 PS3 PM2 | PATH | |
| [81] TN351 | c.1637G>A | p.(R546H) | 1M | FMR1 c.990+14C>T VUS | ? | PM2 | VUS |
| Ref. | Location | Number Affected with Variant, Sex (M/F) | Inheritance in Proband | Patient and/or Functional Data | ACMG Criteria | Conclusion | |
|---|---|---|---|---|---|---|---|
| DNA | r./p. | ||||||
| [69] | “c.-332G>C” (promoter) | 1M (male DD cohort) | Reduced reporter expression to 5.9% of WT (10 hemizygotes in gnomAD, rs922007219) | BS1 BS2 PS3 | VUS | ||
| [69] | “c.-293T>C” (promoter) | 1M (male DD cohort) | Reduced reporter expression to 29.2% of WT(7 hemizygotes in gnomAD, rs1222840333) | BS1 BS2 PS3 | VUS | ||
| [69] | c.-254A>G | 1M (male DD cohort) | Reduced reporter expression to 36.2% of WT(8 hemizygotes in gnomAD, rs1217601043) | BS1 BS2 PS3 | VUS | ||
| [82] | CGG repeat 26 of 31 CGG>CCG | N/A (new EagI site) | 1M | Maternal (unaffected) | Normal % of FMRP+ lymphocytes, but statistically significant decrease to 76% normal FMRP level in lymphoblastoid cell line | None | VUS |
| [83] | c.18G>T | p.(V6=) | 2M (unrelated) | Maternal (unaffected) in 1 | Normal intron 1 splicing in lymphoblastoid cell line (1 patient), normal FMRP Western blot in lymphoblastoid cell line (1 patient) | BS1 BS2 BS3 | BEN |
| [79] | c.18G>T | p.(V6=) | 4/508 male ID/DD cohort | ||||
| [69] | c.18G>T | p.(V6=) | 13 samples, 19 controls | Observed in multiple unaffected controls | |||
| [67] | c.52-1_52delinsTA | 2 abnormal transcripts | 1M + mother | Maternal (affected) | No normal transcripts; 2 abnormal transcripts with skipped exon 2 and skipped exons 2–3; FMRP absent in lymphoblastoid cell line | PVS1 PS3 PP1 | PATH |
| [37] #3 | c.420-8A>G | r.419_420ins420-7_420-1 (p.(M140Ifs*3)) | 1M (ID cohort) | Maternal (unaffected, no XCI skewing) | Cryptic splice acceptor leading to retention of 7 nt from intron 5 (blood) | PS3 PM2 | LPATH |
| [84,85] | c.801G>A (IVS8-1) | r.631_801del (p.(S211_G267del)); exon 8 skipping | 1M (ID cohort) | De novo with maternity/paternity confirmed | No family history of DD (parental first cousin consanguinity) Exon 8 skipping with no normal RT-PCR product; rat model with deletion of exon 8 is affected | PS2 PS3 PM2 | PATH |
| [86] | c.879A>C (IVS9-2) | p.(V293=); reported abnormal splicing intron 9 | 1F (autism/ID cohort) | Inclusion of intron 9 sequence in 23/36 subclones from peripheral blood cDNA | BS2 | VUS | |
| [27] | c.879A>C (IVS9-2) | p.(V293=); no splicing abnormality found | 1M (ID, 1 FXS feature) | Maternal (unaffected) | No abnormal splice amplicons found; normal exon 9–10 junction in RT-PCR product sequence in blood; FMRP present in blood homogenate (1 hemizygote in gnomAD, rs782013865) | ||
| [77] | c.881-1G>T | ? (exon 10 splice acceptor) | 1M (clinical suspicion) | Maternal (1:99 skewed XCI, unaffected) | PVS1 PM2 | LPATH | |
| [37] | c.990+1G>A | r.881_990del (p.(K295Nfs*11)) (exon 10 skipping) | 1M (ID cohort) | De novo | Exon 10 skipping in blood | PVS1 PS3 PM2 PM6 | PATH |
| [69] | c.990+4T>C | ? (intron 10) | 1M (DD cohort) | PM2 | VUS | ||
| [81] | c.990+14C>T | r.881_990del (p.(K295Nfs*11)) (exon 10 skipping) | 3 unrelated males (ID, FXS feature cohort) | Exon 10 skipping on peripheral blood RT-PCR product sequencing in 2 probands (TN-183, TN-351) | BA1 BS2 | BEN | |
| [87] | c.990+14C>T | 81 control individuals | Observed in many controls from general population | ||||
| [79] | c.990+14C>T | 45/508 in ID/DD cohort | |||||
| [88] | c.990+14C>T | 7M/4F among 88 patients with ASD | Statistically significant (p = 0.0123) higher frequency in ASD patients vs. controls Stably inherited in unaffected family members | ||||
| [89] | c.990+14C>T | No significant transmission disequilibrium (p=0.26) Allele frequency 65% (22/34) in East Asian controls; concluded that previously observed association with ASD was false positive due to population stratification (gnomAD AF > 10%, rs25714) | |||||
| [27] | c.*312_313dupT | 1 (ID, 1 FXS feature cohort) | PM2 | VUS | |||
| [69,90] | c.*746T>C | 2M (proband and half-brother from DD cohort) | Faster mRNA decay with FMRP level 80% of normal in lymphoblastoid cell line; variant sufficient and necessary to decrease reporter gene expression in vitro; loss of metabotropic glutamate receptor-stimulated upregulation of reporter expression in transfected mouse neurons (72 hemizygotes in gnomAD, rs183130936) | PS3 PP1 BS1 BS2 | VUS | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tekendo-Ngongang, C.; Grochowsky, A.; Solomon, B.D.; Yano, S.T. Beyond Trinucleotide Repeat Expansion in Fragile X Syndrome: Rare Coding and Noncoding Variants in FMR1 and Associated Phenotypes. Genes 2021, 12, 1669. https://doi.org/10.3390/genes12111669
Tekendo-Ngongang C, Grochowsky A, Solomon BD, Yano ST. Beyond Trinucleotide Repeat Expansion in Fragile X Syndrome: Rare Coding and Noncoding Variants in FMR1 and Associated Phenotypes. Genes. 2021; 12(11):1669. https://doi.org/10.3390/genes12111669
Chicago/Turabian StyleTekendo-Ngongang, Cedrik, Angela Grochowsky, Benjamin D. Solomon, and Sho T. Yano. 2021. "Beyond Trinucleotide Repeat Expansion in Fragile X Syndrome: Rare Coding and Noncoding Variants in FMR1 and Associated Phenotypes" Genes 12, no. 11: 1669. https://doi.org/10.3390/genes12111669
APA StyleTekendo-Ngongang, C., Grochowsky, A., Solomon, B. D., & Yano, S. T. (2021). Beyond Trinucleotide Repeat Expansion in Fragile X Syndrome: Rare Coding and Noncoding Variants in FMR1 and Associated Phenotypes. Genes, 12(11), 1669. https://doi.org/10.3390/genes12111669