
Accurate Single-Cell Clustering through Ensemble Similarity Learning



Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Parameter Settings for Each Algorithm
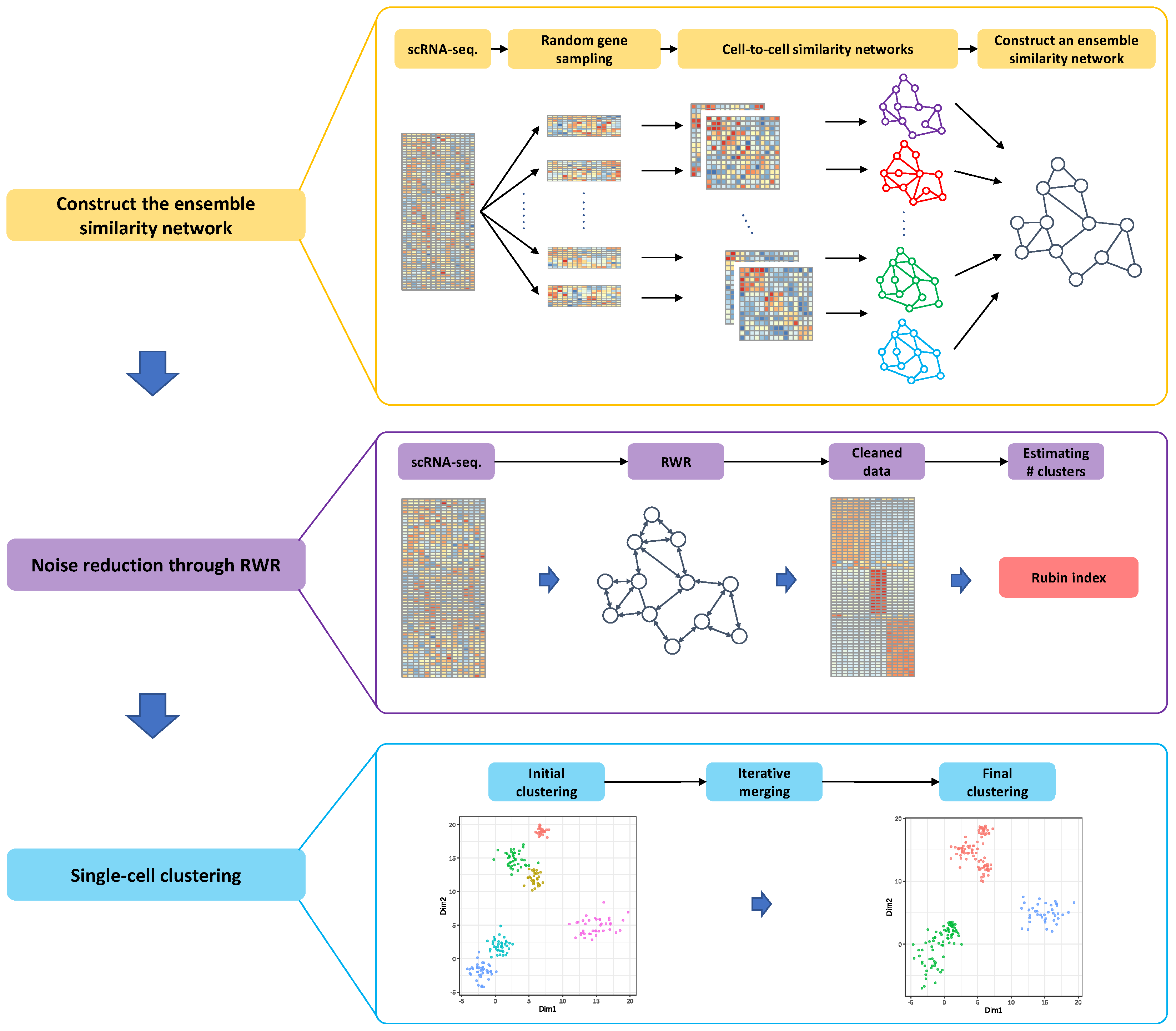
2.3. Motivation and Overview of the Proposed Method
2.4. Data Normalization
2.5. Ensemble Similarity Network Construction
2.6. Reducing Zero-Inflated Noise in a Single-Cell Sequencing Data
2.7. Method to Estimate the True Number of Clusters
2.8. Effective Single-Cell Clustering Algorithm through the Ensemble Similarity Network
| Algorithm 1: Proposed single-cell clustering algorithm. |
 |
2.9. Performance Assessment Metrics
3. Results
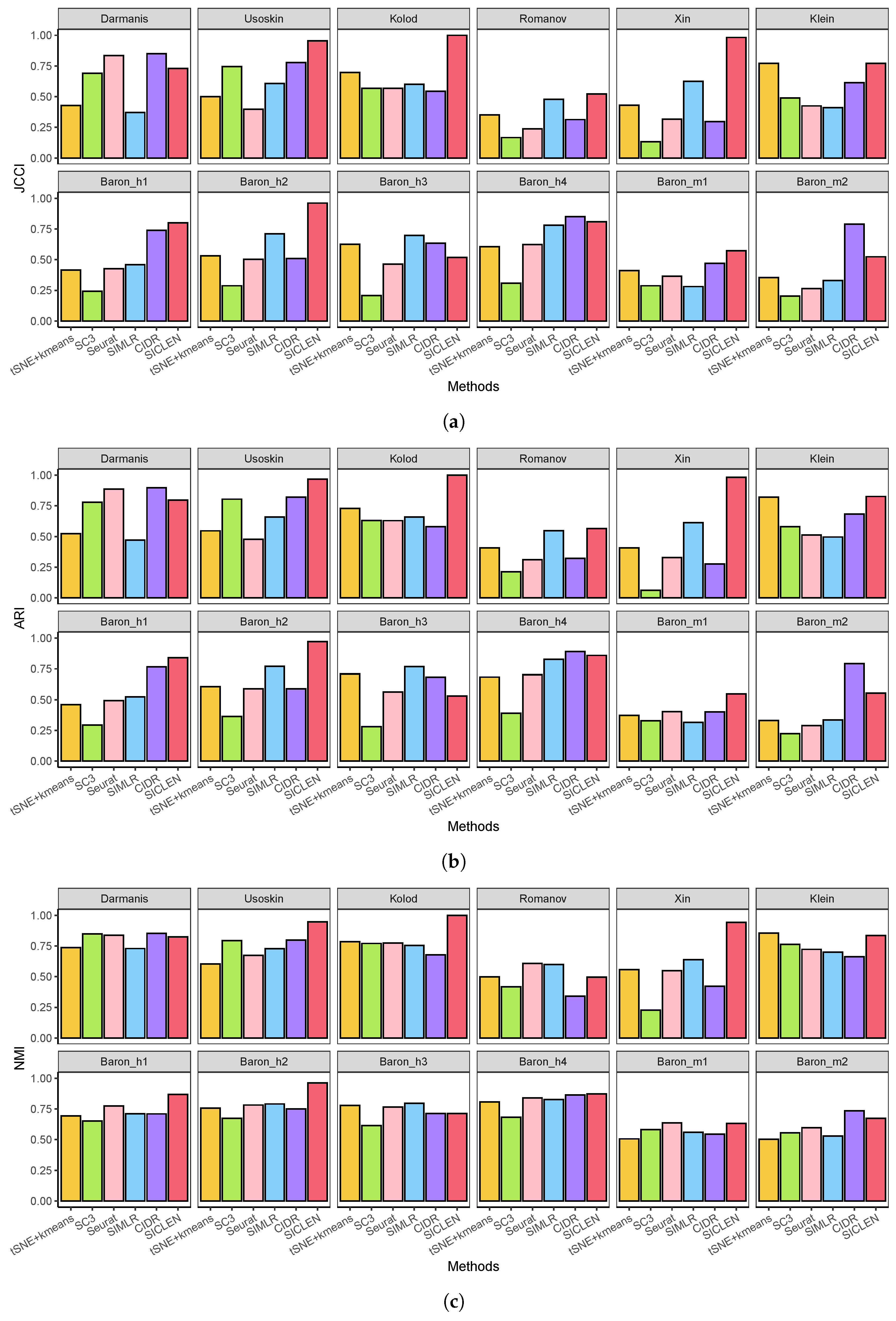
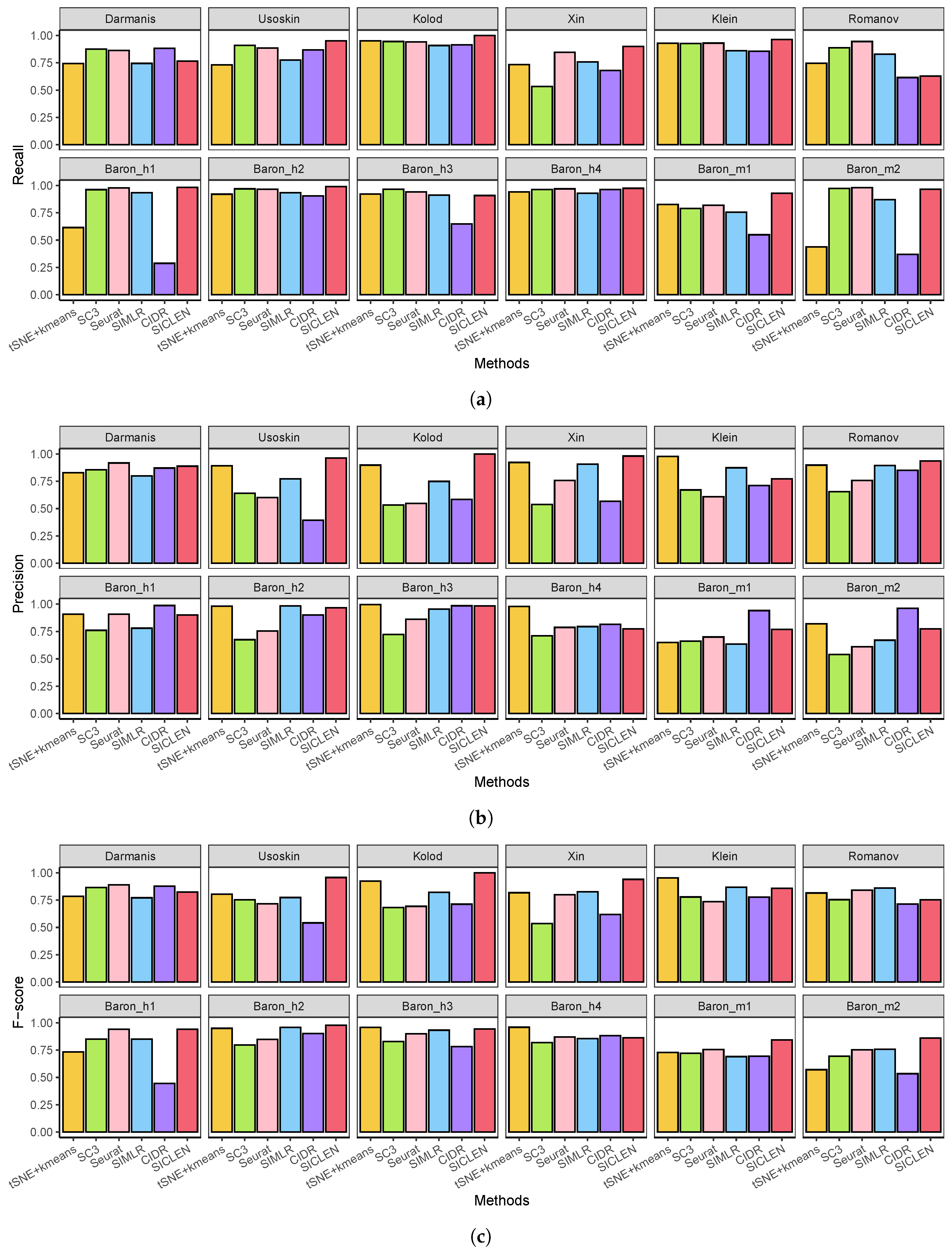
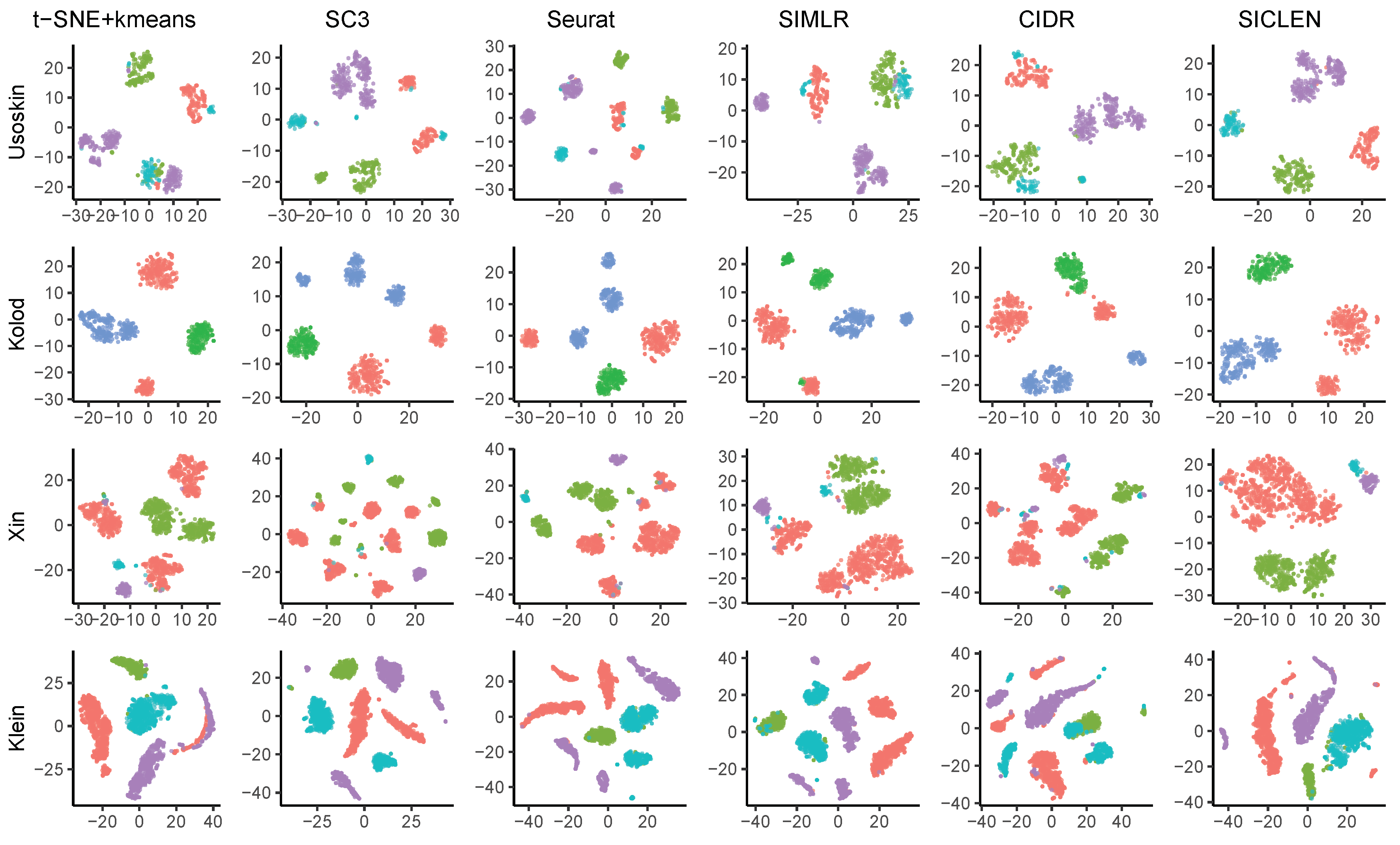
3.1. Performance Assessment Based on the True Cell-Type Labels
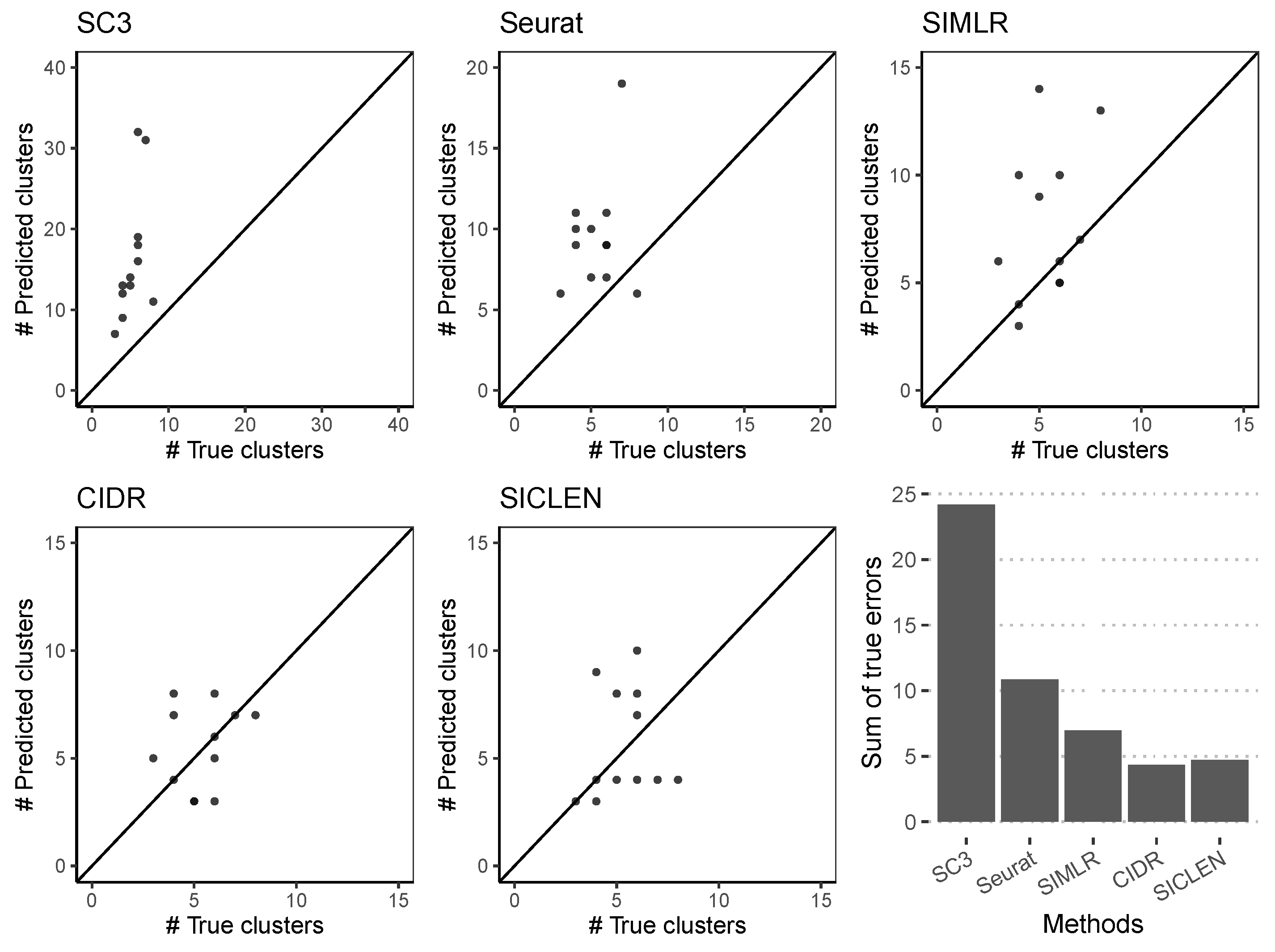
3.2. Accurate Estimation of the True Number of Clusters through an Effective Noise Reduction
3.3. Precise Identification of Differentially Expressed Genes through an Accurate Clustering
3.4. Improved Single-Cell Sequencing Imputation through an Accurate Clustering
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.A.; Kirschner, M.W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015, 161, 1187–1201. [Google Scholar] [CrossRef] [Green Version]
- Islam, S.; Zeisel, A.; Joost, S.; La Manno, G.; Zajac, P.; Kasper, M.; Lönnerberg, P.; Linnarsson, S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods 2014, 11, 163. [Google Scholar] [CrossRef]
- Hashimshony, T.; Wagner, F.; Sher, N.; Yanai, I. CEL-Seq: Single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012, 2, 666–673. [Google Scholar] [CrossRef] [Green Version]
- Luecken, M.D.; Theis, F.J. Current best practices in single-cell RNA-seq analysis: A tutorial. Mol. Syst. Biol. 2019, 15, e8746. [Google Scholar] [CrossRef] [PubMed]
- Yau, C. pcaReduce: Hierarchical clustering of single cell transcriptional profiles. BMC Bioinform. 2016, 17, 140. [Google Scholar]
- Ji, Z.; Ji, H. TSCAN: Pseudo-time reconstruction and evaluation in single-cell RNA-seq analysis. Nucleic Acids Res. 2016, 44, e117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, H.; Khunlertgit, N. Effective single-cell clustering through ensemble feature selection and similarity measurements. Comput. Biol. Chem. 2020, 87, 107283. [Google Scholar] [CrossRef]
- Blondel, V.D.; Guillaume, J.L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008. [Google Scholar] [CrossRef] [Green Version]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Su, Z. Identification of cell types from single-cell transcriptomes using a novel clustering method. Bioinformatics 2015, 31, 1974–1980. [Google Scholar] [CrossRef] [Green Version]
- Levine, J.H.; Simonds, E.F.; Bendall, S.C.; Davis, K.L.; El-ad, D.A.; Tadmor, M.D.; Litvin, O.; Fienberg, H.G.; Jager, A.; Zunder, E.R.; et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 2015, 162, 184–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Zhu, J.; Pierson, E.; Ramazzotti, D.; Batzoglou, S. Visualization and analysis of single-cell RNA-seq data by kernel-based similarity learning. Nat. Methods 2017, 14, 414–416. [Google Scholar] [CrossRef]
- Lin, P.; Troup, M.; Ho, J.W. CIDR: Ultrafast and accurate clustering through imputation for single-cell RNA-seq data. Genome Biol. 2017, 18, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kiselev, V.Y.; Kirschner, K.; Schaub, M.T.; Andrews, T.; Yiu, A.; Chandra, T.; Natarajan, K.N.; Reik, W.; Barahona, M.; Green, A.R.; et al. SC3: Consensus clustering of single-cell RNA-seq data. Nat. Methods 2017, 14, 483–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, R.; Li, M.; Liang, Z.; Wu, F.X.; Pan, Y.; Wang, J. SinNLRR: A robust subspace clustering method for cell type detection by non-negative and low-rank representation. Bioinformatics 2019, 35, 3642–3650. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z. Clustering Single-Cell RNA-Seq Data with Regularized Gaussian Graphical Model. Genes 2021, 12, 311. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.P.; Rubin, J. On some invariant criteria for grouping data. J. Am. Stat. Assoc. 1967, 62, 1159–1178. [Google Scholar] [CrossRef]
- Darmanis, S.; Sloan, S.A.; Zhang, Y.; Enge, M.; Caneda, C.; Shuer, L.M.; Gephart, M.G.H.; Barres, B.A.; Quake, S.R. A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci. USA 2015, 112, 7285–7290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picardi, E.; Horner, D.S.; Pesole, G. Single-cell transcriptomics reveals specific RNA editing signatures in the human brain. RNA 2017, 23, 860–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usoskin, D.; Furlan, A.; Islam, S.; Abdo, H.; Lönnerberg, P.; Lou, D.; Hjerling-Leffler, J.; Haeggström, J.; Kharchenko, O.; Kharchenko, P.V.; et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 2015, 18, 145–153. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Kim, J.K.; Tsang, J.C.; Ilicic, T.; Henriksson, J.; Natarajan, K.N.; Tuck, A.C.; Gao, X.; Bühler, M.; Liu, P.; et al. Single cell RNA-sequencing of pluripotent states unlocks modular transcriptional variation. Cell Stem Cell 2015, 17, 471–485. [Google Scholar] [CrossRef] [Green Version]
- Romanov, R.A.; Zeisel, A.; Bakker, J.; Girach, F.; Hellysaz, A.; Tomer, R.; Alpar, A.; Mulder, J.; Clotman, F.; Keimpema, E.; et al. Molecular interrogation of hypothalamic organization reveals distinct dopamine neuronal subtypes. Nat. Neurosci. 2017, 20, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Kim, J.; Okamoto, H.; Ni, M.; Wei, Y.; Adler, C.; Murphy, A.J.; Yancopoulos, G.D.; Lin, C.; Gromada, J. RNA sequencing of single human islet cells reveals type 2 diabetes genes. Cell Metab. 2016, 24, 608–615. [Google Scholar] [CrossRef] [Green Version]
- Baron, M.; Veres, A.; Wolock, S.L.; Faust, A.L.; Gaujoux, R.; Vetere, A.; Ryu, J.H.; Wagner, B.K.; Shen-Orr, S.S.; Klein, A.M.; et al. A single-cell transcriptomic map of the human and mouse pancreas reveals inter-and intra-cell population structure. Cell Syst. 2016, 3, 346–360. [Google Scholar] [CrossRef] [Green Version]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Pinkert, S.; Schultz, J.; Reichardt, J. Protein interaction networks—More than mere modules. PLoS Comput. Biol. 2010, 6, e1000659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Qian, X. Functional module identification in protein interaction networks by interaction patterns. Bioinformatics 2014, 30, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Charrad, M.; Ghazzali, N.; Boiteau, V.; Niknafs, A. NbClust: An R Package for Determining the Relevant Number of Clusters in a Data Set. J. Stat. Softw. 2014, 61, 1–36. [Google Scholar] [CrossRef] [Green Version]
- Mouselimis, L. ClusterR: Gaussian Mixture Models, K-Means, Mini-Batch-Kmeans, K-Medoids and Affinity Propagation Clustering; R Package Version 1.2.4.; R Foundation for Statistical Compution: Vienna, Austria, 2021. [Google Scholar]
- Finak, G.; McDavid, A.; Yajima, M.; Deng, J.; Gersuk, V.; Shalek, A.K.; Slichter, C.K.; Miller, H.W.; McElrath, M.J.; Prlic, M.; et al. MAST: A flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015, 16, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Song, D.; Li, J.J. PseudotimeDE: Inference of differential gene expression along cell pseudotime with well-calibrated p-values from single-cell RNA sequencing data. Genome Biol. 2021, 22, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Van den Berge, K.; De Bezieux, H.R.; Street, K.; Saelens, W.; Cannoodt, R.; Saeys, Y.; Dudoit, S.; Clement, L. Trajectory-based differential expression analysis for single-cell sequencing data. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Marco, E.; Karp, R.L.; Guo, G.; Robson, P.; Hart, A.H.; Trippa, L.; Yuan, G.C. Bifurcation analysis of single-cell gene expression data reveals epigenetic landscape. Proc. Natl. Acad. Sci. USA 2014, 111, E5643–E5650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, H.; Kiryu, H.; Furusawa, C.; Ko, M.S.; Ko, S.B.; Gouda, N.; Hayashi, T.; Nikaido, I. SCODE: An efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation. Bioinformatics 2017, 33, 2314–2321. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.E.; Stumpf, M.P.; Babtie, A.C. Gene regulatory network inference from single-cell data using multivariate information measures. Cell Syst. 2017, 5, 251–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moerman, T.; Aibar Santos, S.; Bravo González-Blas, C.; Simm, J.; Moreau, Y.; Aerts, J.; Aerts, S. GRNBoost2 and Arboreto: Efficient and scalable inference of gene regulatory networks. Bioinformatics 2019, 35, 2159–2161. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, D.; Sharma, R.; Nainys, J.; Yim, K.; Kathail, P.; Carr, A.J.; Burdziak, C.; Moon, K.R.; Chaffer, C.L.; Pattabiraman, D.; et al. Recovering gene interactions from single-cell data using data diffusion. Cell 2018, 174, 716–729. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Wang, J.; Torre, E.; Dueck, H.; Shaffer, S.; Bonasio, R.; Murray, J.I.; Raj, A.; Li, M.; Zhang, N.R. SAVER: Gene expression recovery for single-cell RNA sequencing. Nat. Methods 2018, 15, 539–542. [Google Scholar] [CrossRef]
- Li, W.V.; Li, J.J. An accurate and robust imputation method scImpute for single-cell RNA-seq data. Nat. Commun. 2018, 9, 997. [Google Scholar] [CrossRef] [Green Version]
- Arisdakessian, C.; Poirion, O.; Yunits, B.; Zhu, X.; Garmire, L.X. DeepImpute: An accurate, fast, and scalable deep neural network method to impute single-cell RNA-seq data. Genome Biol. 2019, 20, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Jeong, H.; Liu, Z. PRIME: A probabilistic imputation method to reduce dropout effects in single-cell RNA sequencing. Bioinformatics 2020, 36, 4021–4029. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | # Genes | # Cells | # Clusters | Source | Accession |
|---|---|---|---|---|---|
| Darmanis [19] | 21,517 | 420 | 8 | Human brain | GSE67835 |
| Usoskin [21] | 19,534 | 622 | 4 | Mouse sensory neurons | GSE59739 |
| Kolod. [22] | 10,684 | 704 | 3 | Mouse embryo stem cells | E-MTAB-2600 |
| Romanov [23] | 21,143 | 2881 | 7 | Mouse hypothalamus | GSE74672 |
| Xin [24] | 33,584 | 1492 | 4 | Human pancreas | GSE81608 |
| Klein [2] | 24,047 | 2717 | 4 | Mouse embryo stem cells | GSE65525 |
| Baron_h1 [25] | 15,452 | 1622 | 6 | Human pancreas | GSE84133 |
| Baron_h2 [25] | 15,810 | 1562 | 6 | Human pancreas | GSE84133 |
| Baron_h3 [25] | 16,386 | 3333 | 6 | Human pancreas | GSE84133 |
| Baron_h4 [25] | 15,285 | 1225 | 6 | Human pancreas | GSE84133 |
| Baron_m1 [25] | 13,757 | 687 | 5 | Mouse pancreas | GSE84133 |
| Baron_m2 [25] | 14,105 | 932 | 5 | Mouse pancreas | GSE84133 |
| Datasets | # DEGs | Datasets | # DEGs |
|---|---|---|---|
| Darmanis | 5828 | Baron_h1 | 695 |
| Usoskin | 2730 | Baron_h2 | 494 |
| Kolod. | 3278 | Baron_h3 | 566 |
| Romanov | 1842 | Baron_h4 | 652 |
| Xin | 3499 | Baron_m1 | 399 |
| Klein | 4819 | Baron_m2 | 527 |
| Method | Darmanis | Usoskin | Kolod. | Xin | Klein | Romanov | Baron_h1 | Baron_h2 | Baron_h3 | Baron_h4 | Baron_m1 | Baron_m2 | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SC3 | 0.633 | 0.608 | 0.127 | 0.593 | 0.379 | 0.721 | 0.564 | 0.631 | 0.646 | 0.537 | 0.799 | 0.831 | 7.068 |
| Seurat | 0.566 | 0.636 | 0.133 | 0.405 | 0.400 | 0.751 | 0.489 | 0.581 | 0.611 | 0.426 | 0.850 | 0.846 | 6.693 |
| SIMLR | 0.729 | 0.605 | 0.163 | 0.344 | 0.387 | 0.665 | 0.479 | 0.557 | 0.551 | 0.376 | 0.873 | 0.788 | 6.515 |
| CIDR | 0.605 | 0.610 | 0.189 | 0.364 | 0.575 | 0.748 | 0.721 | 0.848 | 0.611 | 0.654 | 1.369 | 0.869 | 8.164 |
| SICLEN | 0.485 | 0.603 | 0.015 | 0.314 | 0.354 | 0.769 | 0.503 | 0.597 | 0.535 | 0.421 | 0.833 | 0.869 | 6.299 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, H.; Shin, S.; Yeom, H.-G. Accurate Single-Cell Clustering through Ensemble Similarity Learning. Genes 2021, 12, 1670. https://doi.org/10.3390/genes12111670
Jeong H, Shin S, Yeom H-G. Accurate Single-Cell Clustering through Ensemble Similarity Learning. Genes. 2021; 12(11):1670. https://doi.org/10.3390/genes12111670
Chicago/Turabian StyleJeong, Hyundoo, Sungtae Shin, and Hong-Gi Yeom. 2021. "Accurate Single-Cell Clustering through Ensemble Similarity Learning" Genes 12, no. 11: 1670. https://doi.org/10.3390/genes12111670
APA StyleJeong, H., Shin, S., & Yeom, H. -G. (2021). Accurate Single-Cell Clustering through Ensemble Similarity Learning. Genes, 12(11), 1670. https://doi.org/10.3390/genes12111670