
ORPER: A Workflow for Constrained SSU rRNA Phylogenies


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
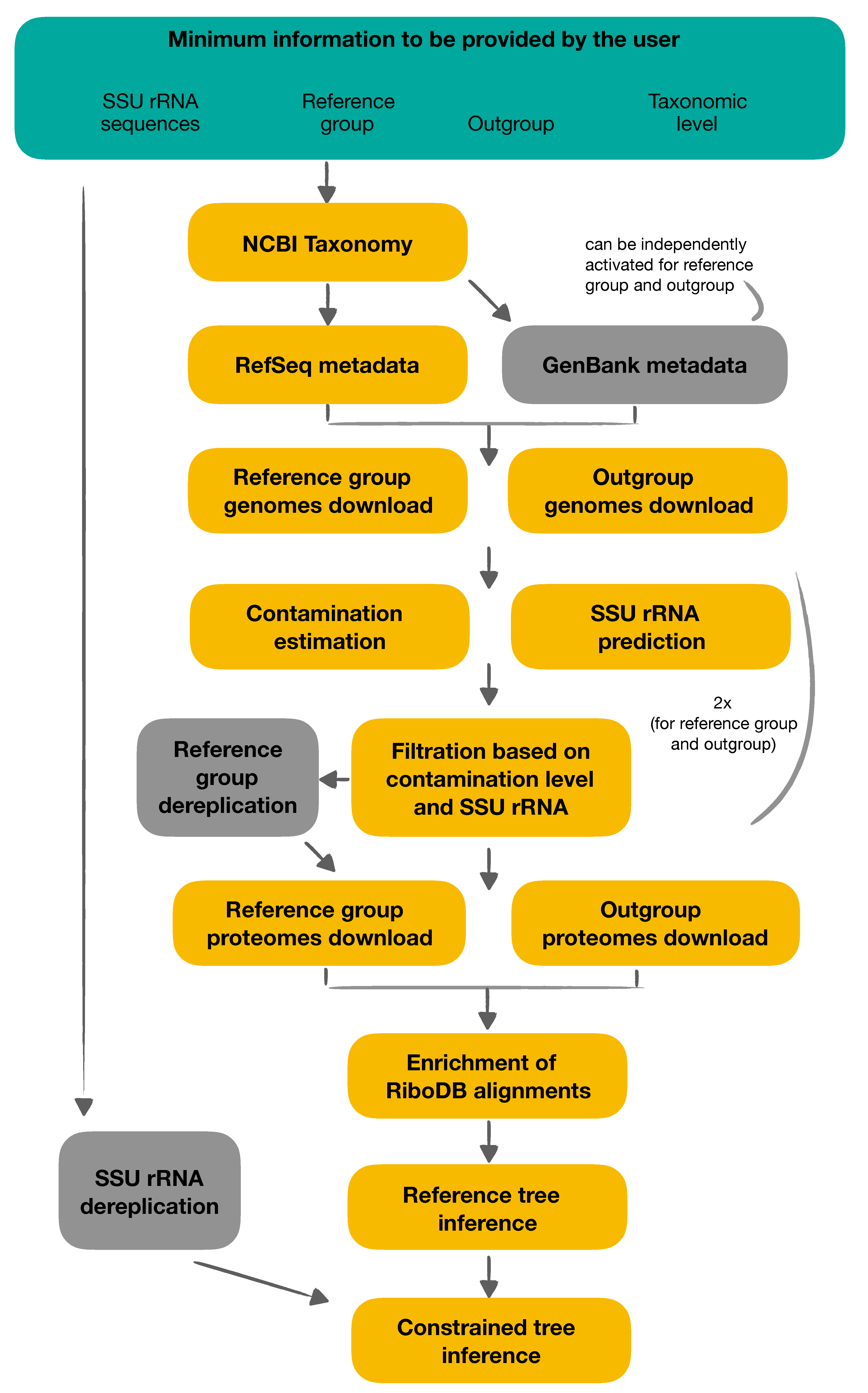
2.1. Functional Overview
2.2. Workflow Details
2.2.1. Taxonomy and Metadata Download
2.2.2. Genome Filtration and Dereplication
2.2.3. Reference Phylogeny Inference
2.2.4. Constrained SSU rRNA Phylogeny
2.3. Design Considerations
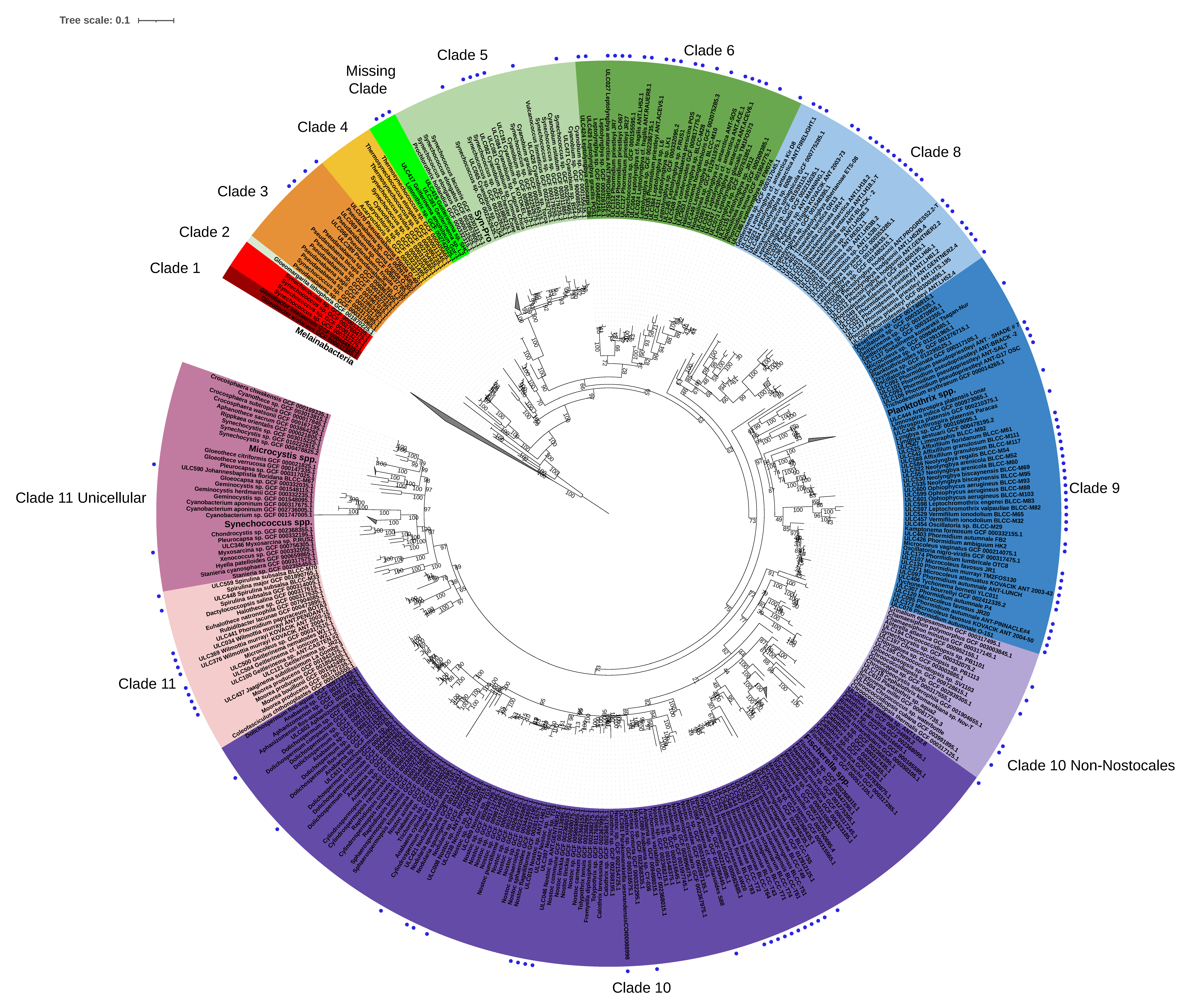
3. Results and Discussion
Case Study: BCCM/ULC Cyanobacteria Collection
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whitton, B.A. Ecology of Cyanobacteria II: Their Diversity in Space and Time; Springer Science & Business Media: Dordrecht, The Netherlands, 2012; ISBN 978-94-007-3855-3. [Google Scholar]
- Ochoa de Alda, J.A.G.; Esteban, R.; Diago, M.L.; Houmard, J. The Plastid Ancestor Originated among One of the Major Cyanobacterial Lineages. Nat. Commun. 2014, 5, 4937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopp, R.E.; Kirschvink, J.L.; Hilburn, I.A.; Nash, C.Z. The Paleoproterozoic Snowball Earth: A Climate Disaster Triggered by the Evolution of Oxygenic Photosynthesis. Proc. Natl. Acad. Sci. USA 2005, 102, 11131–11136. [Google Scholar] [CrossRef] [Green Version]
- Knoll, A.H. The Geological Consequences of Evolution. Geobiology 2003, 1, 3–14. [Google Scholar] [CrossRef]
- Archibald, J.M. The Puzzle of Plastid Evolution. Curr. Biol. 2009, 19, R81–R88. [Google Scholar] [CrossRef] [Green Version]
- Sato, N. Are Cyanobacteria an Ancestor of Chloroplasts or Just One of the Gene Donors for Plants and Algae? Genes 2021, 12, 823. [Google Scholar] [CrossRef]
- Di Rienzi, S.C.; Sharon, I.; Wrighton, K.C.; Koren, O.; Hug, L.A.; Thomas, B.C.; Goodrich, J.K.; Bell, J.T.; Spector, T.D.; Banfield, J.F.; et al. The Human Gut and Groundwater Harbor Non-Photosynthetic Bacteria Belonging to a New Candidate Phylum Sibling to Cyanobacteria. eLife 2013, 2, e01102. [Google Scholar] [CrossRef]
- Mareš, J.; Strunecký, O.; Bučinská, L.; Wiedermannová, J. Evolutionary Patterns of Thylakoid Architecture in Cyanobacteria. Front. Microbiol. 2019, 10, 277. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.R.; Magnabosco, C.; Momper, L.; Gold, D.A.; Bosak, T.; Fournier, G.P. An Expanded Ribosomal Phylogeny of Cyanobacteria Supports a Deep Placement of Plastids. Front. Microbiol. 2019, 10, 1612. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Baracaldo, P.; Cardona, T. On the Origin of Oxygenic Photosynthesis and Cyanobacteria. New Phytol. 2020, 225, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Baracaldo, P.; Bianchini, G.; Wilson, J.D.; Knoll, A.H. Cyanobacteria and Biogeochemical Cycles through Earth History. Trends Microbiol. 2021. [Google Scholar] [CrossRef]
- Chen, M.-Y.; Teng, W.-K.; Zhao, L.; Hu, C.-X.; Zhou, Y.-K.; Han, B.-P.; Song, L.-R.; Shu, W.-S. Comparative Genomics Reveals Insights into Cyanobacterial Evolution and Habitat Adaptation. ISME J. 2021, 15, 211–227. [Google Scholar] [CrossRef]
- Boden, J.S.; Konhauser, K.O.; Robbins, L.J.; Sánchez-Baracaldo, P. Timing the Evolution of Antioxidant Enzymes in Cyanobacteria. Nat. Commun. 2021, 12, 4742. [Google Scholar] [CrossRef]
- Monchamp, M.-E.; Spaak, P.; Pomati, F. Long Term Diversity and Distribution of Non-Photosynthetic Cyanobacteria in Peri-Alpine Lakes. Front. Microbiol. 2019, 9, 3344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, P.M.; Wu, D.; Latifi, A.; Axen, S.D.; Fewer, D.P.; Talla, E.; Calteau, A.; Cai, F.; de Marsac, N.T.; Rippka, R.; et al. Improving the Coverage of the Cyanobacterial Phylum Using Diversity-Driven Genome Sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 1053–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornet, L.; Wilmotte, A.; Javaux, E.J.; Baurain, D. A Constrained SSU-rRNA Phylogeny Reveals the Unsequenced Diversity of Photosynthetic Cyanobacteria (Oxyphotobacteria). BMC Res. Notes 2018, 11, 435. [Google Scholar] [CrossRef] [Green Version]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.-H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the Classification of Cultured and Uncultured Bacteria and Archaea Using 16S rRNA Gene Sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow Enables Reproducible Computational Workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Kurtzer, G.M.; Sochat, V.; Bauer, M.W. Singularity: Scientific Containers for Mobility of Compute. PLoS ONE 2017, 12, e0177459. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; Mcveigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A Comprehensive Update on Curation, Resources and Tools. Database 2020, 2020. [Google Scholar] [CrossRef]
- Haft, D.H.; DiCuccio, M.; Badretdin, A.; Brover, V.; Chetvernin, V.; O’Neill, K.; Li, W.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; et al. RefSeq: An Update on Prokaryotic Genome Annotation and Curation. Nucleic Acids Res. 2018, 46, D851–D860. [Google Scholar] [CrossRef]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. DRep: A Tool for Fast and Accurate Genomic Comparisons That Enables Improved Genome Recovery from Metagenomes through de-Replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jauffrit, F.; Penel, S.; Delmotte, S.; Rey, C.; de Vienne, D.M.; Gouy, M.; Charrier, J.-P.; Flandrois, J.-P.; Brochier-Armanet, C. RiboDB Database: A Comprehensive Resource for Prokaryotic Systematics. Mol. Biol. Evol. 2016, 33, 2170–2172. [Google Scholar] [CrossRef] [Green Version]
- Simion, P.; Philippe, H.; Baurain, D.; Jager, M.; Richter, D.J.; Di Franco, A.; Roure, B.; Satoh, N.; Quéinnec, É.; Ereskovsky, A.; et al. A Large and Consistent Phylogenomic Dataset Supports Sponges as the Sister Group to All Other Animals. Curr. Biol. 2017, 27, 958–967. [Google Scholar] [CrossRef] [Green Version]
- Van Vlierberghe, M.; Di Franco, A.; Philippe, H.; Baurain, D. Decontamination, Pooling and Dereplication of the 678 Samples of the Marine Microbial Eukaryote Transcriptome Sequencing Project. BMC Res. Notes 2021, 14, 306. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A Multiple Sequence Alignment Method with Reduced Time and Space Complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Criscuolo, A.; Gribaldo, S. BMGE (Block Mapping and Gathering with Entropy): A New Software for Selection of Phylogenetic Informative Regions from Multiple Sequence Alignments. BMC Evol. Biol. 2010, 10, 210. [Google Scholar] [CrossRef] [Green Version]
- Roure, B.; Rodriguez-Ezpeleta, N.; Philippe, H. SCaFoS: A Tool for Selection, Concatenation and Fusion of Sequences for Phylogenomics. BMC Evol. Biol. 2007, 7, S2. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML-VI-HPC: Maximum Likelihood-Based Phylogenetic Analyses with Thousands of Taxa and Mixed Models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Gontcharov, A.A.; Marin, B.; Melkonian, M. Are Combined Analyses Better Than Single Gene Phylogenies? A Case Study Using SSU RDNA and RbcL Sequence Comparisons in the Zygnematophyceae (Streptophyta). Mol. Biol. Evol. 2004, 21, 612–624. [Google Scholar] [CrossRef]
- Dessimoz, C.; Gil, M. Phylogenetic Assessment of Alignments Reveals Neglected Tree Signal in Gaps. Genome Biol. 2010, 11, R37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunter, G.; Rocco, A.; Mimouni, N.; Heger, A.; Caldeira, A.; Hein, J. Uncertainty in Homology Inferences: Assessing and Improving Genomic Sequence Alignment. Genome Res. 2008, 18, 298–309. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.M.; Suchard, M.A.; Huelsenbeck, J.P. Alignment Uncertainty and Genomic Analysis. Science 2008, 319, 473–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mareš, J. Multilocus and SSU rRNA Gene Phylogenetic Analyses of Available Cyanobacterial Genomes, and Their Relation to the Current Taxonomic System. Hydrobiologia 2018, 811, 19–34. [Google Scholar] [CrossRef]
- Harrison, P.W.; Ahamed, A.; Aslam, R.; Alako, B.T.F.; Burgin, J.; Buso, N.; Courtot, M.; Fan, J.; Gupta, D.; Haseeb, M.; et al. The European Nucleotide Archive in 2020. Nucleic Acids Res. 2021, 49, D82–D85. [Google Scholar] [CrossRef]
- Evans, J.T.; Denef, V.J. To Dereplicate or Not to Dereplicate? mSphere 2020, 5, e00971-19. [Google Scholar] [CrossRef]
- Cornet, L.; Meunier, L.; Vlierberghe, M.V.; Léonard, R.R.; Durieu, B.; Lara, Y.; Misztak, A.; Sirjacobs, D.; Javaux, E.J.; Philippe, H.; et al. Consensus Assessment of the Contamination Level of Publicly Available Cyanobacterial Genomes. PLoS ONE 2018, 13, e0200323. [Google Scholar] [CrossRef] [Green Version]
- Breitwieser, F.P.; Pertea, M.; Zimin, A.V.; Salzberg, S.L. Human Contamination in Bacterial Genomes Has Created Thousands of Spurious Proteins. Genome Res. 2019, 29, 954–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Minimum Information about a Single Amplified Genome (MISAG) and a Metagenome-Assembled Genome (MIMAG) of Bacteria and Archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirrmeister, B.E.; Antonelli, A.; Bagheri, H.C. The Origin of Multicellularity in Cyanobacteria. BMC Evol. Biol. 2011, 11, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirrmeister, B.E.; de Vos, J.M.; Antonelli, A.; Bagheri, H.C. Evolution of Multicellularity Coincided with Increased Diversification of Cyanobacteria and the Great Oxidation Event. Proc. Natl. Acad. Sci. USA 2013, 110, 1791–1796. [Google Scholar] [CrossRef] [Green Version]
- Urrejola, C.; von Dassow, P.; van den Engh, G.; Salas, L.; Mullineaux, C.W.; Vicuña, R.; Sánchez-Baracaldo, P. Loss of Filamentous Multicellularity in Cyanobacteria: The Extremophile Gloeocapsopsis Sp. Strain UTEX B3054 Retained Multicellular Features at the Genomic and Behavioral Levels. J. Bacteriol. 2021, 2021, e00514-19. [Google Scholar] [CrossRef]
- Oliver, T.; Sánchez-Baracaldo, P.; Larkum, A.W.; Rutherford, A.W.; Cardona, T. Time-Resolved Comparative Molecular Evolution of Oxygenic Photosynthesis. Biochim. Et Biophys. Acta BBA Bioenerg. 2021, 1862, 148400. [Google Scholar] [CrossRef]
- Cardona, T. Thinking Twice about the Evolution of Photosynthesis. Open Biol. 2019, 2019, 180246. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Pichel, F.; Lombard, J.; Soule, T.; Dunaj, S.; Wu, S.H.; Wojciechowski, M.F. Timing the Evolutionary Advent of Cyanobacteria and the Later Great Oxidation Event Using Gene Phylogenies of a Sunscreen. mBio 2021, 10, e00561-19. [Google Scholar] [CrossRef] [Green Version]
- Ponce-Toledo, R.I.; Deschamps, P.; López-García, P.; Zivanovic, Y.; Benzerara, K.; Moreira, D. An Early-Branching Freshwater Cyanobacterium at the Origin of Plastids. Curr. Biol. 2017, 27, 386–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponce-Toledo, R.I.; López-García, P.; Moreira, D. Horizontal and Endosymbiotic Gene Transfer in Early Plastid Evolution. New Phytol. 2019, 224, 618–624. [Google Scholar] [CrossRef] [Green Version]
- Criscuolo, A.; Gribaldo, S. Large-Scale Phylogenomic Analyses Indicate a Deep Origin of Primary Plastids within Cyanobacteria. Mol. Biol. Evol. 2011, 28, 3019–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deusch, O.; Landan, G.; Roettger, M.; Gruenheit, N.; Kowallik, K.V.; Allen, J.F.; Martin, W.; Dagan, T. Genes of Cyanobacterial Origin in Plant Nuclear Genomes Point to a Heterocyst-Forming Plastid Ancestor. Mol. Biol. Evol. 2008, 25, 748–761. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cornet, L.; Ahn, A.-C.; Wilmotte, A.; Baurain, D. ORPER: A Workflow for Constrained SSU rRNA Phylogenies. Genes 2021, 12, 1741. https://doi.org/10.3390/genes12111741
Cornet L, Ahn A-C, Wilmotte A, Baurain D. ORPER: A Workflow for Constrained SSU rRNA Phylogenies. Genes. 2021; 12(11):1741. https://doi.org/10.3390/genes12111741
Chicago/Turabian StyleCornet, Luc, Anne-Catherine Ahn, Annick Wilmotte, and Denis Baurain. 2021. "ORPER: A Workflow for Constrained SSU rRNA Phylogenies" Genes 12, no. 11: 1741. https://doi.org/10.3390/genes12111741
APA StyleCornet, L., Ahn, A.-C., Wilmotte, A., & Baurain, D. (2021). ORPER: A Workflow for Constrained SSU rRNA Phylogenies. Genes, 12(11), 1741. https://doi.org/10.3390/genes12111741