
Genetic Variability of the Functional Domains of Chromodomains Helicase DNA-Binding (CHD) Proteins



 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Retrieval and Alignment of CHD Proteins
2.2. Phylogenetic Analysis of CHD Proteins
2.3. Alignment of CHD Functional Domains
2.4. Retrieval of Pathogenic Missense Variants
3. Results
3.1. Phylogenetic Analysis of CHD Proteins
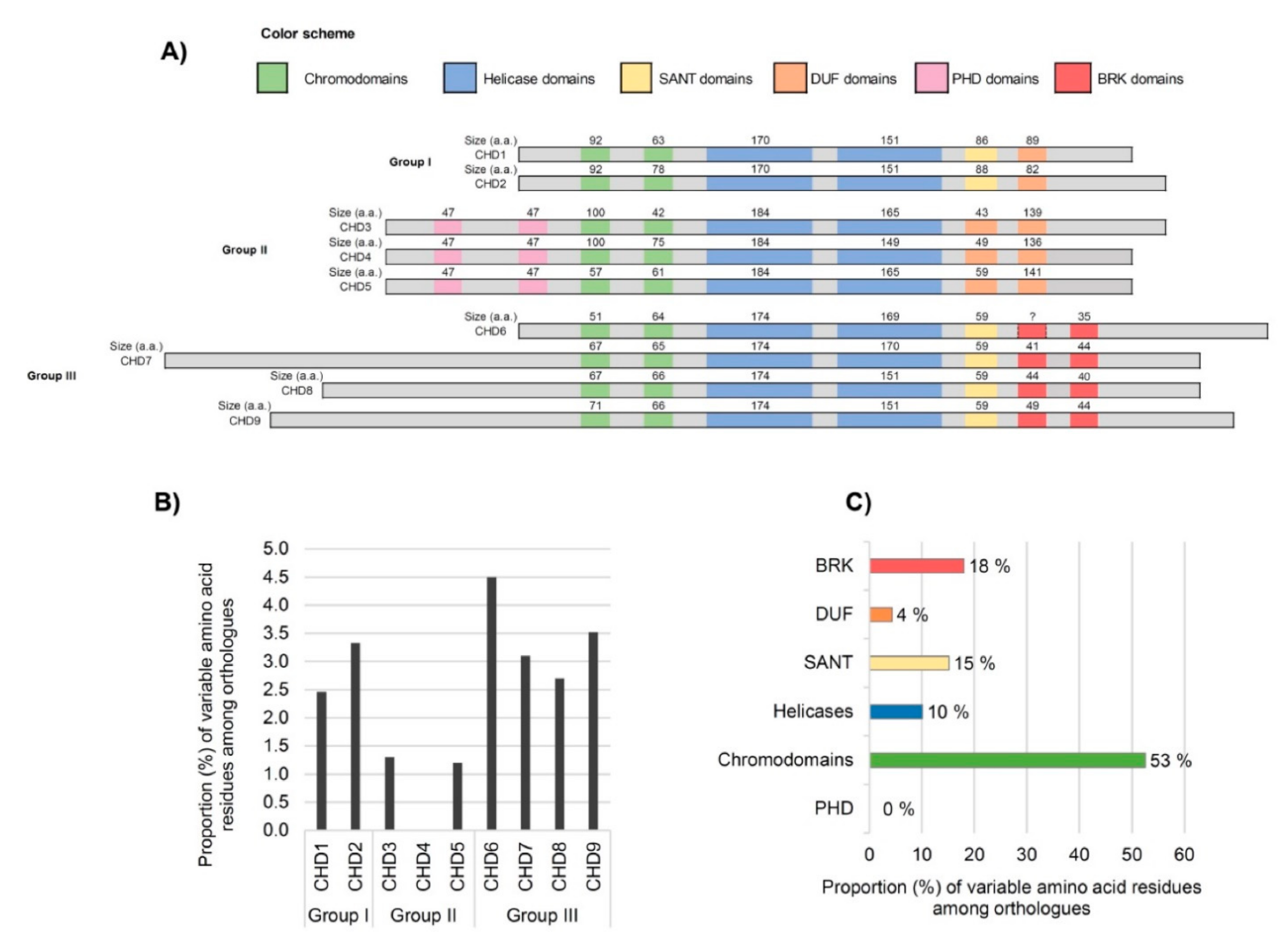
3.2. Analysis of Domains between Human CHDs
3.3. Inter-Species Comparison of CHD Domains
3.4. Analyses of the Pathological Diversity Associated with CHDs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, W.; Mills, A.A. Architects of the genome: Chd dysfunction in cancer, developmental disorders and neurological syndromes. Epigenomics 2014, 6, 381–395. [Google Scholar] [CrossRef] [Green Version]
- Gaspar-Maia, A.; Alajem, A.; Polesso, F.; Sridharan, R.; Mason, M.J.; Heidersbach, A.; Ramalho-Santos, J.; McManus, M.T.; Plath, K.; Meshorer, E.; et al. Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature 2009, 460, 863–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.J.; Khoshkhoo, S.; Frankowski, J.C.; Zhu, B.; Abbasi, S.; Lee, S.; Wu, Y.E.; Hunt, R.F. Chd2 is necessary for neural circuit development and long-term memory. Neuron 2018, 100, 1180–1193.e1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.M. Chromatin remodeling in development and disease: Focus on chd7. PLoS Genet. 2010, 6, e1001010. [Google Scholar] [CrossRef] [PubMed]
- Ostapcuk, V.; Mohn, F.; Carl, S.H.; Basters, A.; Hess, D.; Iesmantavicius, V.; Lampersberger, L.; Flemr, M.; Pandey, A.; Thomä, N.H.; et al. Activity-dependent neuroprotective protein recruits hp1 and chd4 to control lineage-specifying genes. Nature 2018, 557, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.M.; Gotoh, T.; Kok, M.; White, P.S.; Brodeur, G.M. Chd5, a new member of the chromodomain gene family, is preferentially expressed in the nervous system. Oncogene 2003, 22, 1002–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.J.; Yusufzai, T. The ATP-dependent chromatin remodeling enzymes chd6, chd7, and chd8 exhibit distinct nucleosome binding and remodeling activities. J. Biol. Chem. 2017, 292, 11927–11936. [Google Scholar] [CrossRef] [Green Version]
- Layman, W.S.; Hurd, E.A.; Martin, D.M. Chromodomain proteins in development: Lessons from charge syndrome. Clin. Genet. 2010, 78, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [Google Scholar] [CrossRef]
- Hoffmann, A.; Spengler, D. Chromatin remodeling complex nurd in neurodevelopment and neurodevelopmental disorders. Front. Genet. 2019, 10, 682. [Google Scholar] [CrossRef] [Green Version]
- Basta, J.; Rauchman, M. The nucleosome remodeling and deacetylase complex in development and disease. Transl. Res. J. Lab. Clin. Med. 2015, 165, 36–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, H.; Kohnken, R.; Svaren, J. The nucleosome remodeling and deacetylase chromatin remodeling (nurd) complex is required for peripheral nerve myelination. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 1517–1527. [Google Scholar] [CrossRef]
- Marfella, C.G.; Imbalzano, A.N. The chd family of chromatin remodelers. Mutat. Res. 2007, 618, 30–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzman-Ayala, M.; Sachs, M.; Koh, F.M.; Onodera, C.; Bulut-Karslioglu, A.; Lin, C.J.; Wong, P.; Nitta, R.; Song, J.S.; Ramalho-Santos, M. Chd1 is essential for the high transcriptional output and rapid growth of the mouse epiblast. Development 2015, 142, 118–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, T.; Ji, F.; Yuan, Z.; Jiao, J. Chd2 is required for embryonic neurogenesis in the developing cerebral cortex. Stem Cells 2015, 33, 1794–1806. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Gao, S.; Schafer, C.; Colijn, S.; Muthukumar, V.; Griffin, C.T. The chromatin-remodeling enzyme CHD3 plays a role in embryonic viability but is dispensable for early vascular development. PLoS ONE 2020, 15, e0235799. [Google Scholar] [CrossRef]
- Goodman, J.V.; Yamada, T.; Yang, Y.; Kong, L.; Wu, D.Y.; Zhao, G.; Gabel, H.W.; Bonni, A. The chromatin remodeling enzyme Chd4 regulates genome architecture in the mouse brain. Nat. Commun. 2020, 11, 3419. [Google Scholar] [CrossRef]
- Ingram, K.G.; Curtis, C.D.; Silasi-Mansat, R.; Lupu, F.; Griffin, C.T. The nurd chromatin-remodeling enzyme chd4 promotes embryonic vascular integrity by transcriptionally regulating extracellular matrix proteolysis. PLoS Genet. 2013, 9, e1004031. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, T.; Hess, R.A.; Kolla, V.; Higashi, M.; Raabe, T.D.; Brodeur, G.M. CHD5 is required for spermiogenesis and chromatin condensation. Mech. Dev. 2014, 131, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Lathrop, M.J.; Chakrabarti, L.; Eng, J.; Rhodes, C.H.; Lutz, T.; Nieto, A.; Liggitt, H.D.; Warner, S.; Fields, J.; Stöger, R.; et al. Deletion of the Chd6 exon 12 affects motor coordination. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 2010, 21, 130–142. [Google Scholar] [CrossRef] [Green Version]
- Micucci, J.A.; Layman, W.S.; Hurd, E.A.; Sperry, E.D.; Frank, S.F.; Durham, M.A.; Swiderski, D.L.; Skidmore, J.M.; Scacheri, P.C.; Raphael, Y.; et al. CHD7 and retinoic acid signaling cooperate to regulate neural stem cell and inner ear development in mouse models of charge syndrome. Hum. Mol. Genet. 2014, 23, 434–448. [Google Scholar] [CrossRef] [Green Version]
- Katayama, Y.; Nishiyama, M.; Shoji, H.; Ohkawa, Y.; Kawamura, A.; Sato, T.; Suyama, M.; Takumi, T.; Miyakawa, T.; Nakayama, K.I. CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature 2016, 537, 675–679. [Google Scholar] [CrossRef]
- Ooga, M.; Funaya, S.; Hashioka, Y.; Fujii, W.; Naito, K.; Suzuki, M.G.; Aoki, F. Chd9 mediates highly loosened chromatin structure in growing mouse oocytes. Biochem. Biophys. Res. Commun. 2018, 500, 583–588. [Google Scholar] [CrossRef]
- Pilarowski, G.O.; Vernon, H.J.; Applegate, C.D.; Boukas, L.; Cho, M.T.; Gurnett, C.A.; Benke, P.J.; Beaver, E.; Heeley, J.M.; Medne, L.; et al. Missense variants in the chromatin remodeler CHD1 are associated with neurodevelopmental disability. J. Med. Genet. 2018, 55, 561–566. [Google Scholar] [CrossRef]
- Carvill, G.L.; Heavin, S.B.; Yendle, S.C.; McMahon, J.M.; O’Roak, B.J.; Cook, J.; Khan, A.; Dorschner, M.O.; Weaver, M.; Calvert, S.; et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat. Genet. 2013, 45, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Suls, A.; Jaehn, J.A.; Kecskés, A.; Weber, Y.; Weckhuysen, S.; Craiu, D.C.; Siekierska, A.; Djémié, T.; Afrikanova, T.; Gormley, P.; et al. De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am. J. Hum. Genet. 2013, 93, 967–975. [Google Scholar] [CrossRef] [Green Version]
- Snijders Blok, L.; Rousseau, J.; Twist, J.; Ehresmann, S.; Takaku, M.; Venselaar, H.; Rodan, L.H.; Nowak, C.B.; Douglas, J.; Swoboda, K.J.; et al. CHD3 helicase domain mutations cause a neurodevelopmental syndrome with macrocephaly and impaired speech and language. Nat. Commun. 2018, 9, 4619. [Google Scholar] [CrossRef] [Green Version]
- Weiss, K.; Terhal, P.A.; Cohen, L.; Bruccoleri, M.; Irving, M.; Martinez, A.F.; Rosenfeld, J.A.; Machol, K.; Yang, Y.; Liu, P.; et al. De novo mutations in CHD4, an ATP-dependent chromatin remodeler gene, cause an intellectual disability syndrome with distinctive dysmorphisms. Am. J. Hum. Genet. 2016, 99, 934–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sifrim, A.; Hitz, M.P.; Wilsdon, A.; Breckpot, J.; Turki, S.H.; Thienpont, B.; McRae, J.; Fitzgerald, T.W.; Singh, T.; Swaminathan, G.J.; et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 2016, 48, 1060–1065. [Google Scholar] [CrossRef]
- Hittner, H.M.; Hirsch, N.J.; Kreh, G.M.; Rudolph, A.J. Colobomatous microphthalmia, heart disease, hearing loss, and mental retardation–A syndrome. J. Pediatric Ophthalmol. Strabismus 1979, 16, 122–128. [Google Scholar] [CrossRef]
- Jongmans, M.C.; Admiraal, R.J.; van der Donk, K.P.; Vissers, L.E.; Baas, A.F.; Kapusta, L.; van Hagen, J.M.; Donnai, D.; de Ravel, T.J.; Veltman, J.A.; et al. Charge syndrome: The phenotypic spectrum of mutations in the CHD7 gene. J. Med. Genet. 2006, 43, 306–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vissers, L.E.; van Ravenswaaij, C.M.; Admiraal, R.; Hurst, J.A.; de Vries, B.B.; Janssen, I.M.; van der Vliet, W.A.; Huys, E.H.; de Jong, P.J.; Hamel, B.C.; et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 2004, 36, 955–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delahaye, A.; Sznajer, Y.; Lyonnet, S.; Elmaleh-Bergès, M.; Delpierre, I.; Audollent, S.; Wiener-Vacher, S.; Mansbach, A.L.; Amiel, J.; Baumann, C.; et al. Familial CHARGE syndrome because of CHD7 mutation: Clinical intra- and interfamilial variability. Clin. Genet. 2007, 72, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Vuorela, P.; Ala-Mello, S.; Saloranta, C.; Penttinen, M.; Pöyhönen, M.; Huoponen, K.; Borozdin, W.; Bausch, B.; Botzenhart, E.M.; Wilhelm, C.; et al. Molecular analysis of the CHD7 gene in CHARGE syndrome: Identification of 22 novel mutations and evidence for a low contribution of large CHD7 deletions. Genet. Med. Off. J. Am. Coll. Med. Genet. 2007, 9, 690–694. [Google Scholar] [CrossRef] [Green Version]
- An, Y.; Zhang, L.; Liu, W.; Jiang, Y.; Chen, X.; Lan, X.; Li, G.; Hang, Q.; Wang, J.; Gusella, J.F.; et al. De novo variants in the Helicase-C domain of CHD8 are associated with severe phenotypes including autism, language disability and overgrowth. Hum. Genet. 2020, 139, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Bernier, R.; Golzio, C.; Xiong, B.; Stessman, H.A.; Coe, B.P.; Penn, O.; Witherspoon, K.; Gerdts, J.; Baker, C.; Vulto-van Silfhout, A.T.; et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 2014, 158, 263–276. [Google Scholar] [CrossRef] [Green Version]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, B.; Grepo, N.; Thompson, B.L.; Kim, J.; Wang, K.; Evgrafov, O.V.; Lu, W.; Knowles, J.A.; Campbell, D.B. The autism-associated gene chromodomain helicase DNA-binding protein 8 (CHD8) regulates noncoding rnas and autism-related genes. Transl. Psychiatry 2015, 5, e568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eising, E.; Carrion-Castillo, A.; Vino, A.; Strand, E.A.; Jakielski, K.J.; Scerri, T.S.; Hildebrand, M.S.; Webster, R.; Ma, A.; Mazoyer, B.; et al. A set of regulatory genes co-expressed in embryonic human brain is implicated in disrupted speech development. Mol. Psychiatry 2019, 24, 1065–1078. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.G.; Kurth, I.; Lan, F.; Meliciani, I.; Wenzel, W.; Eom, S.H.; Kang, G.B.; Rosenberger, G.; Tekin, M.; Ozata, M.; et al. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and kallmann syndrome. Am. J. Hum. Genet. 2008, 83, 511–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legendre, M.; Abadie, V.; Attié-Bitach, T.; Philip, N.; Busa, T.; Bonneau, D.; Colin, E.; Dollfus, H.; Lacombe, D.; Toutain, A.; et al. Phenotype and genotype analysis of a french cohort of 119 patients with CHARGE syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Lin, Y.; Nery, J.R.; Urich, M.A.; Breschi, A.; Davis, C.A.; Dobin, A.; Zaleski, C.; Beer, M.A.; Chapman, W.C.; et al. Comparison of the transcriptional landscapes between human and mouse tissues. Proc. Natl. Acad. Sci. USA 2014, 111, 17224–17229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, S.; Nagarajan, P.; Wall, J.; Donovan, D.J.; Donell, R.L.; Ligon, A.H.; Venkatachalam, S.; Quade, B.J. Disruption of chromodomain helicase DNA binding protein 2 (CHD2) causes scoliosis. Am. J. Med. Genet. Part A 2008, 146a, 1117–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittaker, D.E.; Riegman, K.L.; Kasah, S.; Mohan, C.; Yu, T.; Pijuan-Sala, B.; Hebaishi, H.; Caruso, A.; Marques, A.C.; Michetti, C.; et al. The chromatin remodeling factor CHD7 controls cerebellar development by regulating reelin expression. J. Clin. Investig. 2017, 127, 874–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sood, S.; Weber, C.M.; Hodges, H.C.; Krokhotin, A.; Shalizi, A.; Crabtree, G.R. CHD8 dosage regulates transcription in pluripotency and early murine neural differentiation. Proc. Natl. Acad. Sci. USA 2020, 117, 22331–22340. [Google Scholar] [CrossRef] [PubMed]
- Alendar, A.; Lambooij, J.P.; Bhaskaran, R.; Lancini, C.; Song, J.Y.; van Vugt, H.; Snoek, M.; Berns, A. Gene expression regulation by the Chromodomain helicase DNA-binding protein 9 (CHD9) chromatin remodeler is dispensable for murine development. PLoS ONE 2020, 15, e0233394. [Google Scholar] [CrossRef]
- The UniProt Consortium. Uniprot: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- NCBI Resource Coordinators. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, J.F.; Blus, B.J.; Kim, D.; Clines, K.L.; Rastinejad, F.; Khorasanizadeh, S. Molecular implications of evolutionary differences in CHD double chromodomains. J. Mol. Biol. 2007, 369, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Criscuolo, A.; Gribaldo, S. BMGE (Block Mapping and Gathering with Entropy): A new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol. 2010, 10, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemoine, F.; Correia, D.; Lefort, V.; Doppelt-Azeroual, O.; Mareuil, F.; Cohen-Boulakia, S.; Gascuel, O. Ngphylogeny.Fr: New generation phylogenetic services for non-specialists. Nucleic Acids Res. 2019, 47, W260–W265. [Google Scholar] [CrossRef] [Green Version]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.Fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed]
- Lefort, V.; Longueville, J.-E.; Gascuel, O. SMS: Smart model selection in phyml. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of phyml 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anisimova, M.; Gil, M.; Dufayard, J.-F.; Dessimoz, C.; Gascuel, O. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst. Biol. 2011, 60, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, J.; Milpetz, F.; Bork, P.; Ponting, C.P. SMART, a simple modular architecture research tool: Identification of signaling domains. Proc. Natl. Acad. Sci. USA 1998, 95, 5857–5864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, N.P.; Leroy, C.; Sander, C. MView: A web-compatible database search or multiple alignment viewer. Bioinformatics 1998, 14, 380–381. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crow, K.D.; Wagner, G.P. What is the role of genome duplication in the evolution of complexity and diversity? Mol. Biol. Evol. 2005, 23, 887–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niklas, K.J.; Newman, S.A. The origins of multicellular organisms. Evol. Dev. 2013, 15, 41–52. [Google Scholar] [CrossRef]
- Trachana, K.; Jensen, L.J.; Bork, P. Evolution and regulation of cellular periodic processes: A role for paralogues. EMBO Rep. 2010, 11, 233–238. [Google Scholar] [CrossRef]
- Goodman, J.V.; Bonni, A. Regulation of neuronal connectivity in the mammalian brain by chromatin remodeling. Curr. Opin. Neurobiol. 2019, 59, 59–68. [Google Scholar] [CrossRef]
- Bosman, E.A.; Penn, A.C.; Ambrose, J.C.; Kettleborough, R.; Stemple, D.L.; Steel, K.P. Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum. Mol. Genet. 2005, 14, 3463–3476. [Google Scholar] [CrossRef]
- Shur, I.; Socher, R.; Benayahu, D. In vivo association of CReMM/CHD9 with promoters in osteogenic cells. J. Cell. Physiol. 2006, 207, 374–378. [Google Scholar] [CrossRef]
- Newton, A.H.; Pask, A.J. CHD9 upregulates RUNX2 and has a potential role in skeletal evolution. BMC Mol. Cell Biol. 2020, 21, 27. [Google Scholar] [CrossRef] [Green Version]
- Eissenberg, J.C. Structural biology of the chromodomain: Form and function. Gene 2012, 496, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.F.; Mi, L.Z.; Chruszcz, M.; Cymborowski, M.; Clines, K.L.; Kim, Y.; Minor, W.; Rastinejad, F.; Khorasanizadeh, S. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature 2005, 438, 1181–1185. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.L.; Zhou, M.M. Structure and mechanisms of lysine methylation recognition by the chromodomain in gene transcription. Biochemistry 2011, 50, 1966–1980. [Google Scholar] [CrossRef] [Green Version]
- Blus, B.J.; Wiggins, K.; Khorasanizadeh, S. Epigenetic virtues of chromodomains. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 507–526. [Google Scholar] [CrossRef] [Green Version]
- Jain, K.; Fraser, C.S.; Marunde, M.R.; Parker, M.M.; Sagum, C.; Burg, J.M.; Hall, N.; Popova, I.K.; Rodriguez, K.L.; Vaidya, A.; et al. Characterization of the plant homeodomain (PHD) reader family for their histone tail interactions. Epigenetics Chromatin 2020, 13, 3. [Google Scholar] [CrossRef]
- Paul, S.; Kuo, A.; Schalch, T.; Vogel, H.; Joshua-Tor, L.; McCombie, W.R.; Gozani, O.; Hammell, M.; Mills, A.A. Chd5 requires PHD-mediated histone 3 binding for tumor suppression. Cell Rep. 2013, 3, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Baker, L.A.; Allis, C.D.; Wang, G.G. PHD fingers in human diseases: Disorders arising from misinterpreting epigenetic marks. Mutat. Res. 2008, 647, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Gonzalez, A.; Calaza, M.; Amigo, J.; González-Peñas, J.; Martínez-Regueiro, R.; Fernández-Prieto, M.; Parellada, M.; Arango, C.; Rodriguez-Fontenla, C.; Carracedo, A. Exploring the biological role of postzygotic and germinal de novo mutations in ASD. Sci. Rep. 2021, 11, 319. [Google Scholar] [CrossRef]
- Ding, H.; Guo, M.; Vidhyasagar, V.; Talwar, T.; Wu, Y. The Q motif is involved in DNA binding but not atp binding in ChlR1 helicase. PLoS ONE 2015, 10, e0140755. [Google Scholar] [CrossRef]
- Weiss, K.; Lazar, H.P.; Kurolap, A.; Martinez, A.F.; Paperna, T.; Cohen, L.; Smeland, M.F.; Whalen, S.; Heide, S.; Keren, B.; et al. The CHD4-related syndrome: A comprehensive investigation of theclinical spectrum, genotype–phenotype correlations, and molecular basis. Genet. Med. 2020, 22, 389–397. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Bergman, J.E.; Bocca, G.; Hoefsloot, L.H.; Meiners, L.C.; van Ravenswaaij-Arts, C.M. Anosmia predicts hypogonadotropic hypogonadism in CHARGE syndrome. J. Pediatrics 2011, 158, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Hale, C.L.; Niederriter, A.N.; Green, G.E.; Martin, D.M. Atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria. Am. J. Med. Genet. Part A 2016, 170a, 344–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalani, S.R.; Safiullah, A.M.; Fernbach, S.D.; Harutyunyan, K.G.; Thaller, C.; Peterson, L.E.; McPherson, J.D.; Gibbs, R.A.; White, L.D.; Hefner, M.; et al. Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. Am. J. Hum. Genet. 2006, 78, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Flaus, A.; Martin, D.M.; Barton, G.J.; Owen-Hughes, T. Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res. 2006, 34, 2887–2905. [Google Scholar] [CrossRef] [Green Version]
- Farnung, L.; Vos, S.M.; Wigge, C.; Cramer, P. Nucleosome-Chd1 structure and implications for chromatin remodelling. Nature 2017, 550, 539–542. [Google Scholar] [CrossRef]
- Yasin, H.; Gibson, W.T.; Langlois, S.; Stowe, R.M.; Tsang, E.S.; Lee, L.; Poon, J.; Tran, G.; Tyson, C.; Wong, C.K.; et al. A distinct neurodevelopmental syndrome with intellectual disability, autism spectrum disorder, characteristic facies, and macrocephaly is caused by defects in CHD8. J. Hum. Genet. 2019, 64, 271–280. [Google Scholar] [CrossRef]
- Rylaarsdam, L.; Guemez-Gamboa, A. Genetic causes and modifiers of autism spectrum disorder. Front. Cell. Neurosci. 2019, 13, 385. [Google Scholar] [CrossRef]
- Pisansky, M.T.; Young, A.E.; O’Connor, M.B.; Gottesman, I.I.; Bagchi, A.; Gewirtz, J.C. Mice lacking the chromodomain helicase DNA-binding 5 chromatin remodeler display autism-like characteristics. Transl. Psychiatry 2017, 7, e1152. [Google Scholar] [CrossRef] [Green Version]
- Moore, S.; Berger, N.D.; Luijsterburg, M.S.; Piett, C.G.; Stanley, F.K.T.; Schräder, C.U.; Fang, S.; Chan, J.A.; Schriemer, D.C.; Nagel, Z.D.; et al. The CHD6 chromatin remodeler is an oxidative DNA damage response factor. Nat. Commun. 2019, 10, 241. [Google Scholar] [CrossRef]
- Mills, A.A. The Chromodomain Helicase DNA-binding chromatin remodelers: Family traits that protect from and promote cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026450. [Google Scholar] [CrossRef] [PubMed]
- Kargapolova, Y.; Rehimi, R.; Kayserili, H.; Brühl, J.; Sofiadis, K.; Zirkel, A.; Palikyras, S.; Mizi, A.; Li, Y.; Yigit, G.; et al. Overarching control of autophagy and DNA damage response by CHD6 revealed by modeling a rare human pathology. Nat. Commun. 2021, 12, 3014. [Google Scholar] [CrossRef] [PubMed]
- Suriano, G.; Azevedo, L.; Novais, M.; Boscolo, B.; Seruca, R.; Amorim, A.; Ghibaudi, E.M. In vitro demonstration of intra-locus compensation using the ornithine transcarbamylase protein as model. Hum. Mol. Genet. 2007, 16, 2209–2214. [Google Scholar] [CrossRef]
- Bridgham, J.T.; Ortlund, E.A.; Thornton, J.W. An epistatic ratchet constrains the direction of glucocorticoid receptor evolution. Nature 2009, 461, 515–519. [Google Scholar] [CrossRef]
- Serrano, C.; Teixeira, C.S.S.; Cooper, D.N.; Carneiro, J.; Lopes-Marques, M.; Stenson, P.D.; Amorim, A.; Prata, M.J.; Sousa, S.F.; Azevedo, L. Compensatory epistasis explored by molecular dynamics simulations. Hum. Genet. 2021, 140, 1329–1342. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Protein | Mutation | Domain | Phenotype | Inheritance | Orthologue Variability | Paralogue Variability | References |
|---|---|---|---|---|---|---|---|
| CHD1 | p.Arg618Gln | Helicase ATP-binding (493-663 a.a.) | Pilarowski-Bjornsson Syndrome (α) | Autosomal Dominant | No | No | Pilarowski et al. (2018) [24] |
| CHD3 | p.Leu915Phe p.His886Asp | Helicase ATP-binding (748–932 a.a.) | Snijders Blok-Campeau Syndrome, Intellectual disability (β) | Autosomal Dominant | No | No | Snijders Blok et al. (2018) [27] |
| p.Arg1121Pro, p.Trp1158Arg p.Asn1159Lys p.Arg1169Trp p.Arg1172Gln | Helicase C-terminal (1064–1229 a.a.) | Snijders Blok-Campeau Syndrome, Intellectual disability (β) | Autosomal Dominant | No | Yes (p.Arg1121Pro) | Snijders Blok et al. (2018) [27] | |
| CHD4 | p.Ser851Tyr | Helicase ATP-binding (738–922 a.a.) | Sifrim-Hitz-Weiss syndrome (γ) | Autosomal Dominant | No | Yes | Sifrim et al. (2016) [29] |
| p.Arg1068His p.Glu1094Lys p.Arg1127Gln p.Trp1148Leu p.Arg1173Leu | Helicase C-terminal (1054–1203 a.a.) | Sifrim-Hitz-Weiss syndrome (γ) | Autosomal Dominant | No | Yes (p.Arg1068His and p.Glu1094Lys) | Sifrim et al. (2016) [29] Weiss et al. (2016) [28] Weiss et al. (2020) [80] Richards et al. (2015) [81] | |
| CHD7 | p.Ser834Phe | Chromo 1 (800–867 a.a.) | CHARGE association, Idiopathic hypogonadotropic hypogonadism (δ) | Autosomal Dominant | No | No | Delahaye et al. (2007) [33] Kim et al. (2008) [41] |
| p.Ile1028Val p.Cys1101Arg | Helicase ATP-binding (980-1154 a.a.) | CHARGE association (δ) | Autosomal Dominant | No | Yes | Vissers et al. (2004) [32] Bergman et al. (2011) [82] | |
| p.Leu1294Pro p.Leu1302Pro | Helicase C-terminal (1294–1464 a.a.) | CHARGE association (δ) | Autosomal Dominant | No | Yes (p.Leu1302Pro) | Hale et al. (2016) [83] Lalani et al. (2006) [84] Legendre et al. (2017) [42] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardoso, A.R.; Lopes-Marques, M.; Oliveira, M.; Amorim, A.; Prata, M.J.; Azevedo, L. Genetic Variability of the Functional Domains of Chromodomains Helicase DNA-Binding (CHD) Proteins. Genes 2021, 12, 1827. https://doi.org/10.3390/genes12111827
Cardoso AR, Lopes-Marques M, Oliveira M, Amorim A, Prata MJ, Azevedo L. Genetic Variability of the Functional Domains of Chromodomains Helicase DNA-Binding (CHD) Proteins. Genes. 2021; 12(11):1827. https://doi.org/10.3390/genes12111827
Chicago/Turabian StyleCardoso, Ana R., Mónica Lopes-Marques, Manuela Oliveira, António Amorim, Maria J. Prata, and Luísa Azevedo. 2021. "Genetic Variability of the Functional Domains of Chromodomains Helicase DNA-Binding (CHD) Proteins" Genes 12, no. 11: 1827. https://doi.org/10.3390/genes12111827
APA StyleCardoso, A. R., Lopes-Marques, M., Oliveira, M., Amorim, A., Prata, M. J., & Azevedo, L. (2021). Genetic Variability of the Functional Domains of Chromodomains Helicase DNA-Binding (CHD) Proteins. Genes, 12(11), 1827. https://doi.org/10.3390/genes12111827