
Remodeling of Neurotransmission, Chemokine, and PI3K-AKT Signaling Genomic Fabrics in Neuropsychiatric Systemic Lupus Erythematosus

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Microarrays
2.3. Filtering and Normalization
2.4. Single Gene Quantifiers
- AVE of each gene in a particular phenotype is expressed in units equal to the AVE of the median gene in all biological replicas of that phenotype.
- REV combines the coefficients of variation (CV) of the expression levels of all spots probing redundantly the same gene in the mid-interval chi-square estimate of the pooled CV.
- Synergistically-expressed genes change their expression levels in phase across biological replicas, antagonistically expressed do so in antiphase, while with independently expressed genes, change of expression of one gene has no direct consequences on the other. From this perspective, the upstream activator and downstream activated genes of a functional pathway should be synergistically expressed with the central gene. By contrast, the upstream inhibitor and downstream inhibited genes should be antagonistically expressed with the central gene. Independently expressed genes should not be considered as related to a pathway.
- If a gene is probed by a single spot (most cases) then two genes are p < 0.05 significantly synergistically expressed if COR > 0.95, significantly antagonistically expressed if COR < −0.95, and independently expressed if −0.025 < COR < 0.025. The absolute cut-off for significant synergism/antagonism changes with more spots redundantly probing the same transcript: |COR| > 0.707 for two spots, …, |COR| > 0.273 for 13 (maximum) number of spots [12].
- COR was determined by the Anaconda distribution of the Python 3 software “CORRELATION”, described in Reference [13].
2.5. Gene Hierarchy and Gene Master Regulator of the Phenotype
2.6. Expression Regulation
2.7. Analysis of the Genomic Fabrics of Functional Pathways
3. Results
3.1. Three Independent Characteristics for Every Gene
3.2. Three Measures of Expression Level Regulation of Individual Genes
3.3. Regulation of Signaling Pathways and NPSLE Biomarkers
3.4. Gene Hierarchy
3.5. Remodeling of the Transcriptomic Networks
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kello, N.; Anderson, E.; Diamond, B. Cognitive dysfunction in systemic lupus erythematosus: A case for initiating trials. Arthritis Rheumatol. 2019, 71, 1413–1425. [Google Scholar] [CrossRef]
- Liang, M.H.; Corzillius, M.; Bae, S.; Lew, R.A.; Fortin, P.R.; Gordon, C.; Isenberg, D.; Alarcon, G.; Straaton, K.V.; Denburg, J.; et al. The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheumatol. 1999, 42, 599–608. [Google Scholar] [CrossRef]
- Schwartz, N.; Stock, A.D.; Putterman, C. Neuropsychiatric lupus: New mechanistic insights and future treatment directions. Nat. Rev. Rheumatol. 2019, 15, 137–152. [Google Scholar] [CrossRef]
- Du, Y.; Sanam, S.; Kate, K.; Mohan, C. Animal models of lupus and lupus nephritis. Curr. Pharm. Des. 2015, 21, 2320–2349. [Google Scholar] [CrossRef] [PubMed]
- Ballok, D.A. Neuroimmunopathology in a murine model of neuropsychiatric lupus. Brain Res. Rev. 2007, 54, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Desplat-Jégo, S.; Varriale, S.; Creidy, R.; Terra, R.; Bernard, D.; Khrestchatisky, M.; Izui, S.; Chicheportiche, Y.; Boucraut, J. TWEAK is expressed by glial cells, induces astrocyte proliferation and increases EAE severity. J. Neuroimmunol. 2002, 133, 116–123. [Google Scholar] [CrossRef]
- Stock, A.D.; Wen, J.; Putterman, C. Neuropsychiatric lupus, the blood brain barrier, and the TWEAK/Fn14 pathway. Front. Immunol. 2013, 4, 484. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Xia, Y.; Stock, A.; Michaelson, J.S.; Burkly, L.C.; Gulinello, M.; Putterman, C. Neuropsychiatric disease in murine lupus is dependent on the TWEAK/Fn14 pathway. J. Autoimmun. 2013, 43, 44–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fragoso-Loyo, H.; Atisha-Fregoso, Y.; Nuñez-Alvarez, C.A.; Llorente, L. Utility of TWEAK to assess neuropsychiatric disease activity in systemic lupus erhytematosus. Lupus 2015, 25, 364–369. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Iacobas, S.; Stout, R.; Spray, D.C. Cellular environment remodels the genomic fabrics of functional pathways in astrocytes. Genes 2020, 11, 520. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, D.A. Biomarkers, master regulators and genomic fabric remodeling in a case of papillary thyroid carcinoma. Genes 2020, 11, 1030. [Google Scholar] [CrossRef]
- Testing the Significance of Pearson’s r. Available online: https://webstat.une.edu.au/unit_materials/c6_common_statistical_tests/test_signif_pearson.html (accessed on 9 December 2020).
- Iacobas, S.; Ede, N.; Iacobas, D.A. The Gene Master Regulators (GMR) approach provides legitimate targets for personalized, time-sensitive cancer gene therapy. Genes 2019, 10, 560. [Google Scholar] [CrossRef] [Green Version]
- Iacobas, D.A.; Iacobas, S.; Lee, P.R.; Cohen, J.E.; Fields, R.D. Coordinated activity of transcriptional networks responding to the pattern of action potential firing in neurons. Genes 2019, 10, 754. [Google Scholar] [CrossRef] [Green Version]
- Arriens, C.; Wren, J.D.; Munroe, M.E.; Mohan, C. Systemic lupus erythematosus biomarkers: The challenging quest. Rheumatology 2016, 56, i32–i45. [Google Scholar] [CrossRef] [Green Version]
- Makinde, H.M.; Winter, D.R.; Procissi, D.; Mike, E.V.; Stock, A.D.; Kando, M.J.; Gadhvi, G.T.; Droho, S.; Bloomfield, C.L.; Dominguez, S.T.; et al. A Novel microglia-specific transcriptional signature correlates with behavioral deficits in neuropsychiatric lupus. Front. Immunol. 2020, 11, 230. [Google Scholar] [CrossRef] [Green Version]
- Iacobas, D.A.; Tuli, N.; Iacobas, S.; Rasamny, J.K.; Moscatello, A.; Geliebter, J.; Tiwari, R.M. Gene master regulators of papillary and anaplastic thyroid cancer phenotypes. Oncotarget 2018, 9, 2410–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, R.; Huang, J.; Iacobas, S.; Iacobas, D.A. Pulmonary hypertension remodels the genomic fabrics of major functional pathways. Genes 2020, 11, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobas, S.; Iacobas, D.A. Astrocyte proximity modulates the myelination gene fabric of oligodendrocytes. Neuron Glia Biol. 2010, 6, 157–169. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- KEGG. Chemokine Signaling Pathway. Available online: https://www.kegg.jp/kegg-bin/show_pathway?mmu04062 (accessed on 9 February 2020).
- KEGG. PI3K-Akt Signaling Pathway. Available online: https://www.genome.jp/kegg-bin/show_pathway?mmu04151 (accessed on 9 February 2020).
- KEGG. Derived Glutamatergic Synapse. Available online: https://www.kegg.jp/kegg-bin/show_pathway?mmu04724 (accessed on 9 February 2020).
- KEGG. Derived GABAergic Synapse. Available online: https://www.kegg.jp/kegg-bin/show_pathway?hsa04727 (accessed on 9 February 2020).
- KEGG. Derived Cholinergic Synapse. Available online: https://www.kegg.jp/kegg-bin/show_pathway?hsa04725 (accessed on 9 February 2020).
- KEGG. Derived Dopaminergic Synpase. Available online: https://www.kegg.jp/kegg-bin/show_pathway?hsa04728 (accessed on 9 February 2020).
- KEGG. Derived Serotonergic Synapse. Available online: https://www.kegg.jp/kegg-bin/show_pathway?hsa04726 (accessed on 9 February 2020).
- Mackay, M.; Vo, A.; Tang, C.C.; Small, M.; Anderson, E.; Ploran, E.J.; Storbeck, J.; Bascetta, B.; Kang, S.; Aranow, C.; et al. Metabolic and microstructural alterations in the SLE brain correlate with cognitive impairment. JCI Insight 2019, 4, e124002. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Kovoor, A.; Nordman, S.; Abu Seman, N.; Gu, T.; Efendic, S.; Brismar, K.; Östenson, C.-G.; Gu, H.F. Increased expression of adenylyl cyclase 3 in pancreatic islets and central nervous system of diabetic Goto-Kakizaki rats. Islets 2012, 4, 343–348. [Google Scholar] [CrossRef] [Green Version]
- Magro-Checa, C.; Steup-Beekman, G.M.; Huizinga, T.W.; Van Buchem, M.A.; Ronen, I. Laboratory and neuroimaging biomarkers in neuropsychiatric systemic lupus erythematosus: Where do we stand, where to go? Front. Med. 2018, 5. [Google Scholar] [CrossRef]
- Davenport, E.E.; Amariuta, T.; Gutierrez-Arcelus, M.; Slowikowski, K.; Westra, H.-J.; Luo, Y.; Shen, C.; Rao, D.A.; Zhang, Y.; Pearson, S.; et al. Discovering in vivo cytokine-eQTL interactions from a lupus clinical trial. Genome Biol. 2018, 19, 168. [Google Scholar] [CrossRef] [Green Version]
- Fortin, S.P.; Ennis, M.J.; Savitch, B.A.; Carpentieri, D.; McDonough, W.S.; Winkles, J.A.; Loftus, J.C.; Kingsley, C.; Hostetter, G.; Tran, N.L. Tumor necrosis factor–like weak inducer of apoptosis stimulation of glioma cell survival is dependent on Akt2 function. Mol. Cancer Res. 2009, 7, 1871–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobas, D.A.; Iacobas, S.; Spray, D.C. Connexin-dependent transcellular transcriptomic networks in mouse brain. Prog. Biophys. Mol. Biol. 2007, 94, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Iacobas, D.A.; Zhou, D.; Chen, Q.; Gavrialov, O.; Haddad, G.G. Gene expression and phenotypic characterization of mouse heart after chronic constant and intermittent hypoxia. Physiol. Genom. 2005, 22, 292–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobas, D.A.; Iacobas, S.; Urban-Maldonado, M.; Scemes, E.; Spray, D.C. Similar transcriptomic alterations in Cx43 knockdown and knockout astrocytes. Cell Commun. Adhes. 2008, 15, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Iacobas, D.A.; Iacobas, S.; Werner, P.; Scemes, E.; Spray, D.C. Alteration of transcriptomic networks in adoptive-transfer experimental autoimmune encephalomyelitis. Front. Integr. Neurosci. 2007, 1, 10. [Google Scholar] [CrossRef] [Green Version]
- Doerner, J.L.; Wen, J.; Xia, Y.; Paz, K.B.; Schairer, D.; Wu, L.; Chalmers, S.A.; Izmirly, P.; Michaelson, J.S.; Burkly, L.C.; et al. TWEAK/Fn14 signaling involvement in the pathogenesis of cutaneous disease in the MRL/lpr model of spontaneous lupus. J. Investig. Dermatol. 2015, 135, 1986–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelson, J.S.; Wisniacki, N.; Burkly, L.C.; Putterman, C. Role of TWEAK in lupus nephritis: A bench-to-bedside review. J. Autoimmun. 2012, 39, 130–142. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef]
- Yang, B.; Yan, P.; Gong, H.; Zuo, L.; Shi, Y.; Guo, J.; Guo, R.; Xie, J.; Li, B. TWEAK protects cardiomyocyte against apoptosis in a PI3K/AKT pathway dependent manner. Am. J. Transl. Res. 2016, 8, 3848–3860. [Google Scholar]
- Dogra, C.; Changotra, H.; Wedhas, N.; Qin, X.; Wergedal, J.E.; Kumar, A. TNF-related weak inducer of apoptosis (TWEAK) is a potent skeletal muscle-wasting cytokine. FASEB J. 2007, 21, 1857–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Xu, M.; Yin, X.; Guo, H.; Zhang, B.; Wang, Y.; Xiao, J.; Zou, X.; Zhang, M.; Zhuge, Y. TWEAK promotes hepatic stellate cell migration through activating EGFR/Src and PI3K/AKT pathways. Cell Biol. Int. 2019. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.-D.; Feng, F.; Yu, X.-S.; Liu, Z.-D.; Lao, L.-F. miR-149-5p inhibits cell growth by regulating TWEAK/Fn14/PI3K/AKT pathway and predicts favorable survival in human osteosarcoma. Int. J. Immunopathol. Pharmacol. 2018, 32, 2058738418786656. [Google Scholar] [CrossRef]
- Tinahones, F.J.; C-Soriguer, F.J.; Mancha, I.; Esteva, I. Malignant paraganglioma with aortic infiltration cured by radical surgery. Ann. Med. Interna 1996, 13, 303. [Google Scholar] [PubMed]
- Bishop, G.A.; Berbari, N.F.; Lewis, J.; Mykytyn, K. Type III adenylyl cyclase localizes to primary cilia throughout the adult mouse brain. J. Comp. Neurol. 2007, 505, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Luo, J.; Leng, Y.; Yang, Y.; Zweifel, L.S.; Palmiter, R.D.; Storm, D.R. Ablation of type III adenylyl cyclase in mice causes reduced neuronal activity, altered sleep pattern, and depression-like phenotypes. Biol. Psychiatry 2016, 80, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Speliotes, E.K.; Willer, C.J.; Berndt, S.I.; Monda, K.L.; Thorleifsson, G.; Jackson, A.U.; Allen, H.L.; Lindgren, C.M.; Luan, J.; Mägi, R.; et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 2010, 42, 937–948. [Google Scholar] [CrossRef] [Green Version]
- Warrington, N.; Howe, L.D.; Paternoster, L.; Kaakinen, M.; Herrala, S.; Huikari, V.; Wu, Y.Y.; Kemp, J.P.; Timpson, N.J.; Pourcain, B.S.; et al. A genome-wide association study of body mass index across early life and childhood. Int. J. Epidemiol. 2015, 44, 700–712. [Google Scholar] [CrossRef] [Green Version]
- Grarup, N.; Moltke, I.; Andersen, M.K.; Dalby, M.; Vitting-Seerup, K.; Kern, T.; Mahendran, Y.; Jørsboe, E.; Larsen, C.V.L.; Dahl-Petersen, I.K.; et al. Loss-of-function variants in ADCY3 increase risk of obesity and type 2 diabetes. Nat. Genet. 2018, 50, 172–174. [Google Scholar] [CrossRef]
- Gonzales, M.M.; Tarumi, T.; Miles, S.C.; Tanaka, H.; Shah, F.; Haley, A.P. Insulin sensitivity as a mediator of the relationship between BMI and Working memory-related brain activation. Obesity 2010, 18, 2131–2137. [Google Scholar] [CrossRef]
- Su, F.; Shu, H.; Ye, Q.; Wang, Z.; Xie, C.; Yuan, B.; Zhang, Z.; Bai, F. Brain insulin resistance deteriorates cognition by altering the topological features of brain networks. Neuroimage Clin. 2017, 13, 280–287. [Google Scholar] [CrossRef]
- Zou, J.; Wu, K.; Lin, C.; Jie, Z.-G. LINC00319 acts as a microRNA-335–5p sponge to accelerate tumor growth and metastasis in gastric cancer by upregulating ADCY3. Am. J. Physiol. Liver Physiol. 2020, 318, G10–G22. [Google Scholar] [CrossRef]
- Pullabhatla, V.; Roberts, A.L.; Lewis, M.J.; Mauro, D.; Morris, D.L.; Odhams, C.A.; Tombleson, P.; Liljedahl, U.; Vyse, S.; Simpson, M.A.; et al. De novo mutations implicate novel genes in systemic lupus erythematosus. Hum. Mol. Genet. 2018, 27, 421–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakzad, B.; Shirpour, R.; Mousavi, M.; Karimzadeh, H.; Salehi, A.; Kazemi, M.; Amini, G.; Akbari, M.; Salehi, R. C1QTNF4 gene p.His198Gln mutation is correlated with early-onset systemic lupus erythematosus in Iranian patients. Int. J. Rheum. Dis. 2020, 23, 1594–1598. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, D.A.; Iacobas, S.; Thomas, N.; Spray, D.C. Sex-dependent gene regulatory networks of the heart rhythm. Funct. Integr. Genom. 2010, 10, 73–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, N.M.; Jasmin, J.F.; Lisanti, M.P.; Iacobas, D.A. Sex differences in expression and subcellular localization of heart rhythm determinant proteins. Biochem. Biophys. Res. Commun. 2011, 406, 117–122. [Google Scholar] [CrossRef]








{kind=link}
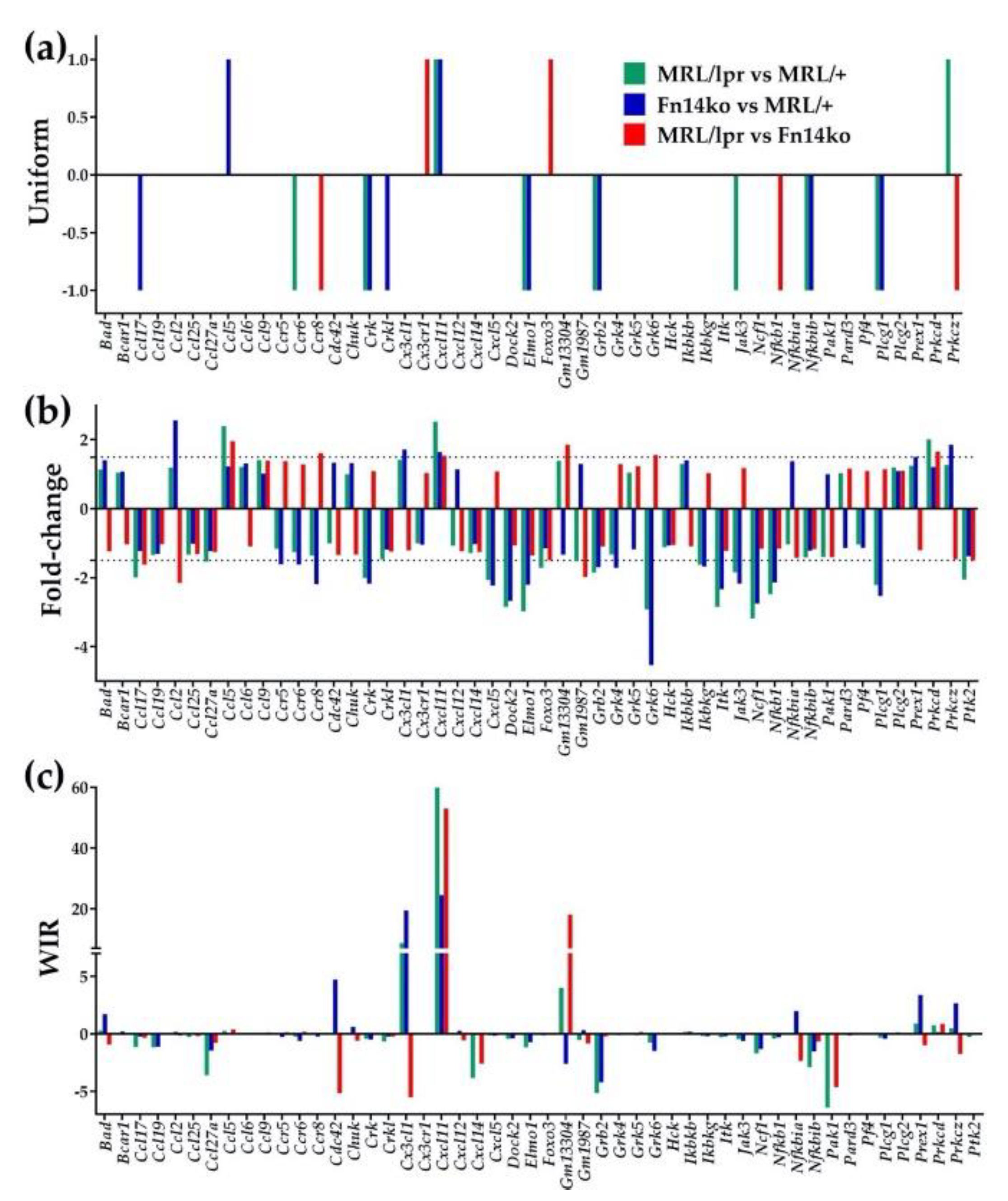{kind=link}
{kind=link}
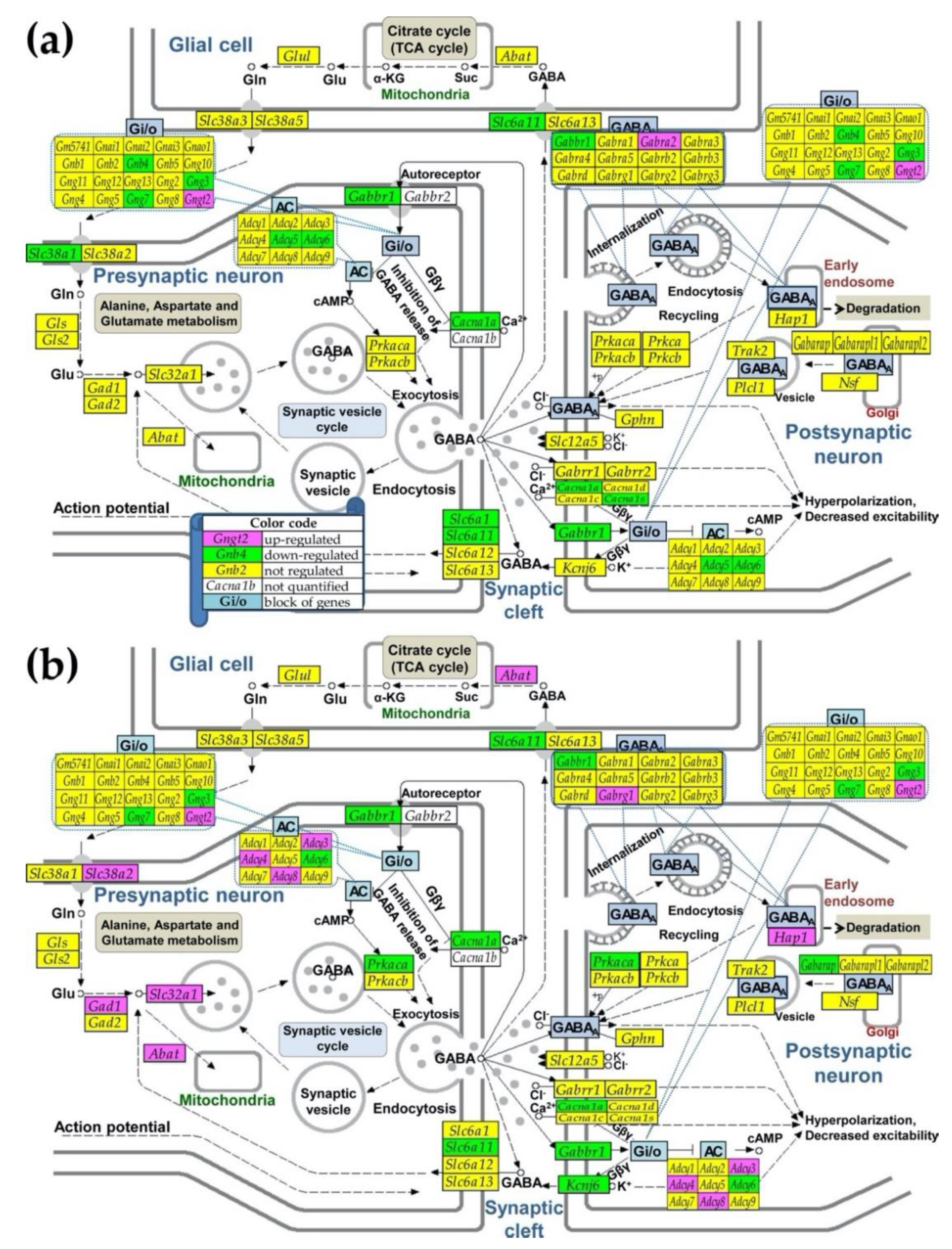{kind=link}
{kind=link}
{kind=link}
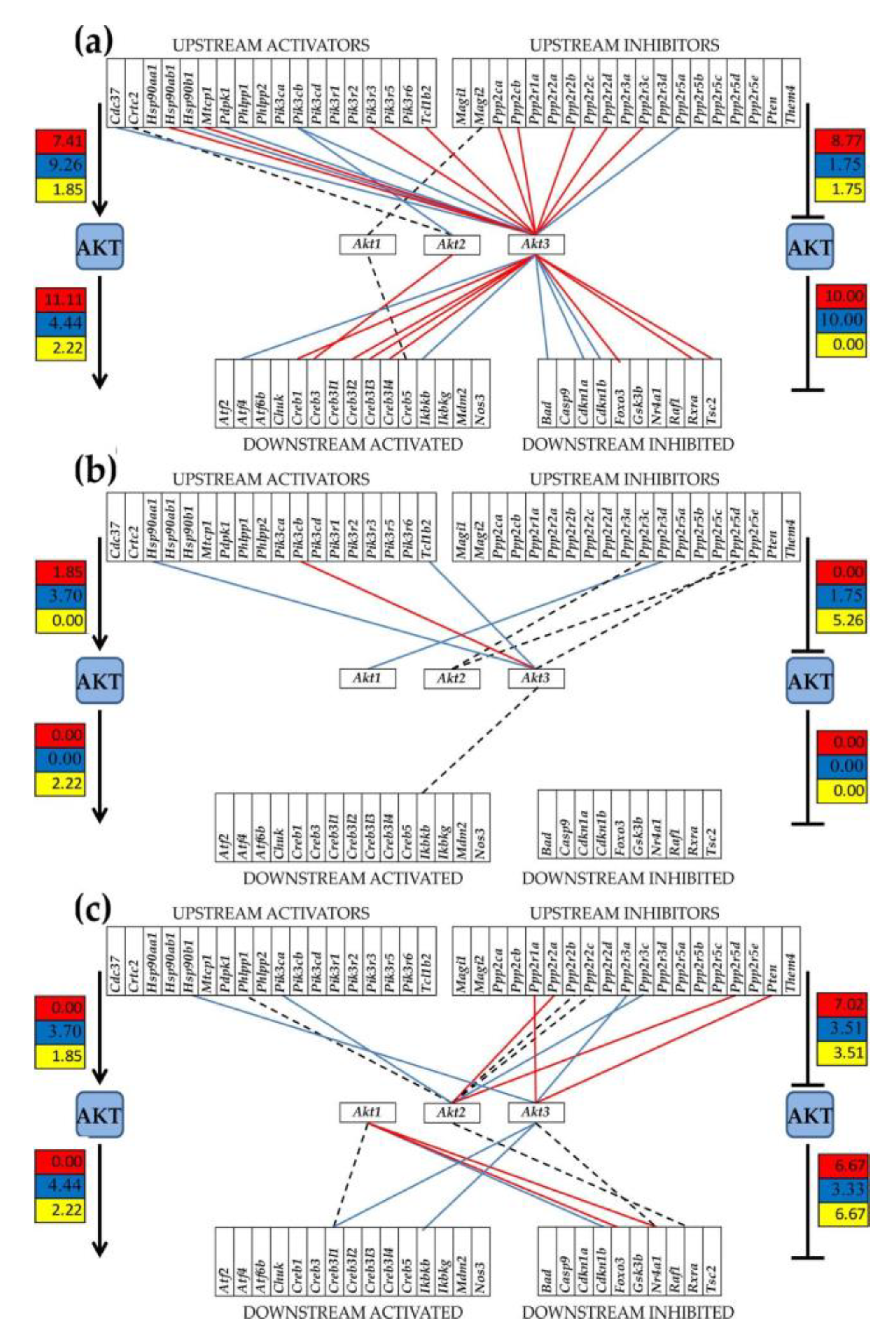{kind=link}
| Gene | Synapse | Description | MRL/lpr | Fn14ko | ||
|---|---|---|---|---|---|---|
| × | WIR | × | WIR | |||
| Adcy3 | CHO,GAB,GLU | adenylate cyclase 3 | 18.95 | 165.74 | 1.22 | 3.22 |
| Adcy5 | CHO,DOP,GAB,GLU,SER | adenylate cyclase 5 | −2.00 | −1.75 | ||
| Adcy6 | CHO,GAB,GLU | adenylate cyclase 6 | −1.89 | −19.08 | −1.67 | −14.38 |
| Akt2 | CHO,DOP | thymoma viral proto-oncogene 2 | 11.01 | 55.62 | ||
| Aft4 | CHO,DOP | activating transcription factor 4 | 2.52 | 11.21 | ||
| Braf | SER | Braf transforming gene | −3.47 | −0.83 | −4.46 | −1.18 |
| Cacna1a | CHO,DOP,GAB,GLU,SER | calcium channel, voltage-dependent, P/Q type, alpha 1A subunit | −1.84 | −1.37 | −3.72 | −4.48 |
| Cacna1s | CHO,GAB,SER | calcium channel, voltage-dependent, L type, alpha 1S subunit | −2.48 | −0.54 | ||
| Calm14 | DOP | calmodulin-like 4 | 1.62 | 29.72 | ||
| Camk2d | CHO,DOP | calcium/calmodulin-dependent protein kinase II, delta | −2.24 | −0.92 | ||
| Chrm1 | CHO | cholinergic receptor, muscarinic 1, CNS | −1.60 | −1.22 | −1.53 | −1.08 |
| Comt | DOP | catechol-O-methyltransferase | −1.83 | −1.75 | ||
| Creb3 | CHO,DOP | cAMP responsive element binding protein 3 | −1.79 | −1.77 | ||
| Cyp2d11 | SER | cytochrome P450, family 2, subfamily d, polypeptide 11 | −1.62 | −2.52 | −1.68 | −2.79 |
| Cyp2d12 | SER | cytochrome P450, family 2, subfamily d, polypeptide 12 | −1.61 | −0.27 | −1.70 | −0.32 |
| Fyn | CHO | Fyn proto-oncogene | −1.38 | −0.85 | −1.37 | −0.83 |
| Gabbr1 | GAB | gamma-aminobutyric acid (GABA) B receptor, 1 | −1.74 | −2.63 | −2.16 | −4.17 |
| Gabra2 | GAB | gamma-aminobutyric acid type A receptor subunit alpha 2 | 1.56 | 1.25 | ||
| Gabrg1 | GAB | gamma-aminobutyric acid type A receptor subunit gamma 1 | 2.07 | 0.73 | ||
| Gm2436 | DOP | Predicted gene 2436 | 1.57 | 19.88 | ||
| Gnal | DOP | guanine nucleotide binding protein, alpha stimulating, olfactory type | 2.14 | 7.00 | 1.73 | 4.41 |
| Gng3 | CHO,DOP,GAB,GLU,SER | guanine nucleotide binding protein (G protein), gamma 3 | −1.53 | −30.02 | −1.21 | −11.35 |
| Gng7 | CHO,DOP,GAB,GLU,SER | guanine nucleotide binding protein (G protein), gamma 7 | −2.07 | −0.56 | −2.53 | −0.81 |
| Gngt2 | CHO,DOP,GAB,GLU,SER | guanine nucleotide binding protein (G protein), gamma transducing activity polypeptide 2 | 1.49 | 0.20 | ||
| Gria2 | DOP,GLU | glutamate receptor, ionotropic, AMPA2 (alpha 2) | −1.52 | −2.69 | ||
| Grin2b | DOP,GLU | glutamate receptor, ionotropic, NMDA2B (epsilon 2) | 1.56 | 0.15 | ||
| Grm2 | GLU | glutamate receptor, metabotropic 2 | −1.87 | −3.10 | −2.28 | −4.56 |
| Grm4 | GLU | glutamate receptor, metabotropic 4 | −3.08 | −2.98 | −2.90 | −2.72 |
| Homer1 | GLU | homer homolog 1 (Drosophila) | −1.66 | −0.63 | ||
| Kcnd2 | SER | potassium voltage-gated channel, Shal-related family, member 2 | 1.30 | 1.32 | ||
| Kras | CHO,SER | v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog | −1.62 | −0.27 | −1.51 | −0.23 |
| Mapk11 | DOP | mitogen-activated protein kinase 11 | −2.37 | −0.45 | −2.50 | −0.50 |
| Mapk12 | DOP | mitogen-activated protein kinase 12 | −2.96 | −3.22 | −1.92 | −1.49 |
| Pik3ca | CHO | phosphatidylinositol 3-kinase, catalytic, alpha polypeptide | −1.95 | −0.89 | −2.32 | −1.23 |
| Pik3cb | CHO | phosphatidylinositol 3-kinase, catalytic, beta polypeptide | −1.15 | −0.25 | −1.75 | −1.26 |
| Plcb3 | CHO,DOP,GLU,SER | phospholipase C, beta 3 | 1.25 | 0.28 | 1.42 | 0.45 |
| Plcb4 | CHO,DOP,GLU,SER | phospholipase C, beta 4 | −2.34 | −1.21 | −1.68 | −0.63 |
| Ppp1cb | DOP | protein phosphatase 1, catalytic subunit, beta isoform | −2.13 | −1.69 | ||
| Ppp1cc | DOP | protein phosphatase 1, catalytic subunit, gamma isoform | −2.02 | −0.88 | −1.39 | −0.33 |
| Ppp2r2a | DOP | protein phosphatase 2 (formerly 2A), regulatory subunit B | −1.89 | −0.57 | −1.88 | −0.56 |
| Ppp2r2c | DOP | protein phosphatase 2 (formerly 2A), regulatory subunit B (PR 52), gamma isoform | −1.85 | −0.64 | −1.44 | −0.32 |
| Ppp2r5e | DOP | protein phosphatase 2, regulatory subunit B (B56), epsilon isoform | −1.80 | −0.42 | −1.60 | −0.32 |
| Rapgef3 | SER | Rap guanine nucleotide exchange factor (GEF) 3 | 1.37 | 0.94 | 1.68 | 1.77 |
| Shank1 | GLU | Rap guanine nucleotide exchange factor (GEF) 3 | −2.96 | −0.88 | −2.32 | −0.58 |
| Slc17a7 | GLU | solute carrier family 17 (sodium-dependent inorganic phosphate cotransporter), member 7 | −2.43 | −44.12 | −2.19 | −36.79 |
| Slc17a8 | GLU | solute carrier family 17 (sodium-dependent inorganic phosphate cotransporter), member 8 | −1.42 | −0.24 | −1.57 | −0.33 |
| Slc38a1 | GAB,GLU | solute carrier family 38, member 1 | −1.32 | −3.57 | ||
| Slc6a1 | GAB | solute carrier family 6 (neurotransmitter transporter, GABA), member 1 | −2.35 | −0.76 | ||
| Slc6a11 | GAB | solute carrier family 6 (neurotransmitter transporter, GABA), member 11 | −2.55 | −6.66 | −2.72 | −7.35 |
| Th | DOP | tyrosine hydroxylase | −2.86 | −0.67 | −2.40 | −0.50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iacobas, D.; Wen, J.; Iacobas, S.; Schwartz, N.; Putterman, C. Remodeling of Neurotransmission, Chemokine, and PI3K-AKT Signaling Genomic Fabrics in Neuropsychiatric Systemic Lupus Erythematosus. Genes 2021, 12, 251. https://doi.org/10.3390/genes12020251
Iacobas D, Wen J, Iacobas S, Schwartz N, Putterman C. Remodeling of Neurotransmission, Chemokine, and PI3K-AKT Signaling Genomic Fabrics in Neuropsychiatric Systemic Lupus Erythematosus. Genes. 2021; 12(2):251. https://doi.org/10.3390/genes12020251
Chicago/Turabian StyleIacobas, Dumitru, Jing Wen, Sanda Iacobas, Noa Schwartz, and Chaim Putterman. 2021. "Remodeling of Neurotransmission, Chemokine, and PI3K-AKT Signaling Genomic Fabrics in Neuropsychiatric Systemic Lupus Erythematosus" Genes 12, no. 2: 251. https://doi.org/10.3390/genes12020251
APA StyleIacobas, D., Wen, J., Iacobas, S., Schwartz, N., & Putterman, C. (2021). Remodeling of Neurotransmission, Chemokine, and PI3K-AKT Signaling Genomic Fabrics in Neuropsychiatric Systemic Lupus Erythematosus. Genes, 12(2), 251. https://doi.org/10.3390/genes12020251