1. Introduction
Adipocytes (classically known for the storage of excess fat and lipids) have now emerged as an active endocrine organ [
1]. In addition to it, there is an increased level of hormones like leptin, resistin, and cytokines in obese subjects compared to non-obese individuals, which cumulatively relates obesity to various co-morbidities [
2,
3,
4]. Surprisingly, not all obese people show increased adipokines levels, and instead, show a healthy profile like a normal-weight individual. The state of obesity showing an absence of the metabolic complication and BMI greater than 30 kg/m
2 has been named as metabolically healthy obesity (MHO) [
5]. On the other hand, the metabolic abnormality can be observed in normal-weight individuals who showed a risk of development of metabolic syndrome with their BMI lying within a healthy normal range. These individuals are called as metabolically unhealthy normal-weight (MUNW) [
6].
Though numerous studies have been published related to metabolic health, and many are undergoing; still, no single defined criteria to predict metabolic health is available [
7]. Alternatively, Karelis put forward an idea of the initial set of simple biochemical estimations as markers to identify the MHO individual based on their research studies and suggested if four out of five metabolic markers are in the normal range, a person can be considered as MHO [
8]. The five metabolic markers in their study were triglycerides (TG), HDL-Cholesterol (HDL-C), LDL-cholesterol (LDL-C), total cholesterol (TC), and insulin resistance.
Dietary pattern, physical inactivity, and a sedentary lifestyle contribute to obesity, though; one should also embark on the influence of genes in the body [
9]. The genetic predisposition to obesity has a vital role in the development of obesity subtypes. Some genes, like
FTO and
SH2B1, are genetically thrifty genes, known to be naturally selected for obesity [
10,
11,
12]. On the other hand, the genetic contribution of pathways involved in adipose tissues also informs us about the development of MHO and MUO phenotypes.
Therefore, taking into account simple biochemical markers, we planned our study with the criteria of MHO proposed by Karelis, 2004, with some modifications [
8]. In the present study, we aimed to analyze the prevalence of metabolically healthy overweight/obesity (MHOW) and metabolically unhealthy normal-weight (MUNW) individuals among young, physically active Indian males belonging to the same region, ethnicity, age, and dietary habits. Moreover, understanding the metabolic profile of such individuals is obscure and requires to be explored with more research; therefore, whole-body composition analyses with hematological parameters and biochemical estimations of hormones and inflammatory markers along with whole-genome expression profiling have been studied in this research article. Since genetic insight into MHOW and MUNW is not studied on physically active individuals yet, this makes our study as distinct and novel.
2. Materials and Methods
2.1. Subjects
The study was conducted on young male volunteers (age ≥ 18) of a fitness center in Karnal, Haryana. Participants were indulged in exercise for a minimum tenure of 6 months and were moderately active. They were explained with the study protocol approved by the Institutional Ethics Committee (IEC DIPAS, IEC/DIPAS/C-1/2 DATED 26.5.15). A total of 120 individuals consented in written for participation in the study. Dietary intake was assessed using a 7-day self-filled dietary intake form. These contained different food items as study participants were free living. Although this method suffers limitations as both over- and under-reporting of food intake are known problems [
13,
14], calorie and nutrient intake was computed from the dietary recalls based on values reported for Indian recipes using the database of National Institute of Nutrition (
http://218.248.6.43:8080/CountWhatYouEat/, accessed on 1 July 2020) and dietary manual (
https://www.nin.res.in/downloads/DietaryGuidelinesforNINwebsite.pdf, accessed on 1 July 2020) [
15]). Out of 120 participants, 24 male participants (six participants per group) with an average age of 23.8 years were picked up for further microarray analysis. Healthy volunteers with a minimum 18 years of age, engaged in more than six months of regular exercise constituted inclusion criteria, while individuals with long term medication, due to acute or chronic illness, were excluded from the study.
The Criterion for Metabolic syndrome (Karelis, 2004, with some modifications) [
8]:
Fasting TG level ≥ 150 mg/dL
Fasting HDL-C ≤ 40 mg/dL (men)
Fasting LDL-C ≥ 100 mg/dL
Fasting TC ≥ 200 mg/dL
C-reactive protein ≥ 1.18 (90th percentile)
Insulin resistance (McAuley’s Index) ≥ 15.76 (90th percentile)
Thus, taking BMI ≥ 24.9 kg/m2 as overweight/obese and the above-mentioned metabolic syndrome, we divided the population into four groups considering zero metabolic syndromes for metabolically health profiles.
Categorization of participants:
Metabolically healthy normal-weight (MHNW): BMI ≤ 24.9 kg/m2, with no markers of metabolic syndrome
Metabolically unhealthy normal-weight (MUNW): BMI ≤ 24.9 kg/m2, with one or more markers of metabolic syndrome
Metabolically healthy overweight/obese (MHOW): BMI ≥ 25 kg/m2, with no markers of metabolic syndrome
Metabolically unhealthy overweight/obese (MUOW): BMI ≥ 25 kg/m2, with one or more markers of metabolic syndrome
2.2. Anthropometric Measurements
Height was measured using a measuring rod with the least count as 0.1 cm (Seca 216, Seca Asia Pacific medical measuring systems and scales, Kuala Lumpur, Malaysia). Bodyweight was measured with bare feet, in light clothing using a bioelectric impedance analyzer (Tanita BC-420MA, body composition analyzer, Tanita Corporation, Tokyo, Japan). Body mass index (BMI) was calculated using the formula weight in kilograms divided by squared height in meters, i.e., Weight (kg)/Height (m2). Body composition analysis was done before breakfast between 0700 h and 1000 h in the post-absorptive state.
2.3. TEE, PAL Value, and BMR Calculation
Total energy expenditure (TEE) for physical activity was monitored using accelerometry-based wearable Actical
® devices (Respironics, mini mitter co. Inc., Bend, OR, USA). The volunteers wore the device on the wrist for 7 days continuously to record minute by minute energy expenditure [
16]. BMR was calculated manually using the prediction equation for Indians [
17]) (
Supplementary Table S1). Further, physical activity level (PAL) was calculated by the division of total energy expenditure (TEE) by the basal metabolic rate (BMR) (PAL = TEE/BMR).
2.4. Collection of Blood Samples
Whole venous blood was collected in plain EDTA and heparinized vacutainers by venipuncture in the arm. Hematological parameters were measured on a fully automatic hematology analyzer (MS4e, MeletSchloesing Lab., Osny, France). Plasma was collected after centrifugation of heparinized whole blood sample for 10 min at 3000 rpm, while serum was isolated after keeping the whole blood sample at room temperature for about 1 h. Serum samples were utilized for evaluating hormones and biochemical variables, while human plasma was conserved with a protease inhibitor for immunoblot analysis. Both the samples were stored at −80 °C. For microarray of whole-genome genes, 2.5 mL of whole blood was drawn by venous puncture directly into PAXgen blood RNA tubes (BD, Franklin Lakes, NJ, USA) to stabilize RNA and stored at −80 °C for further use.
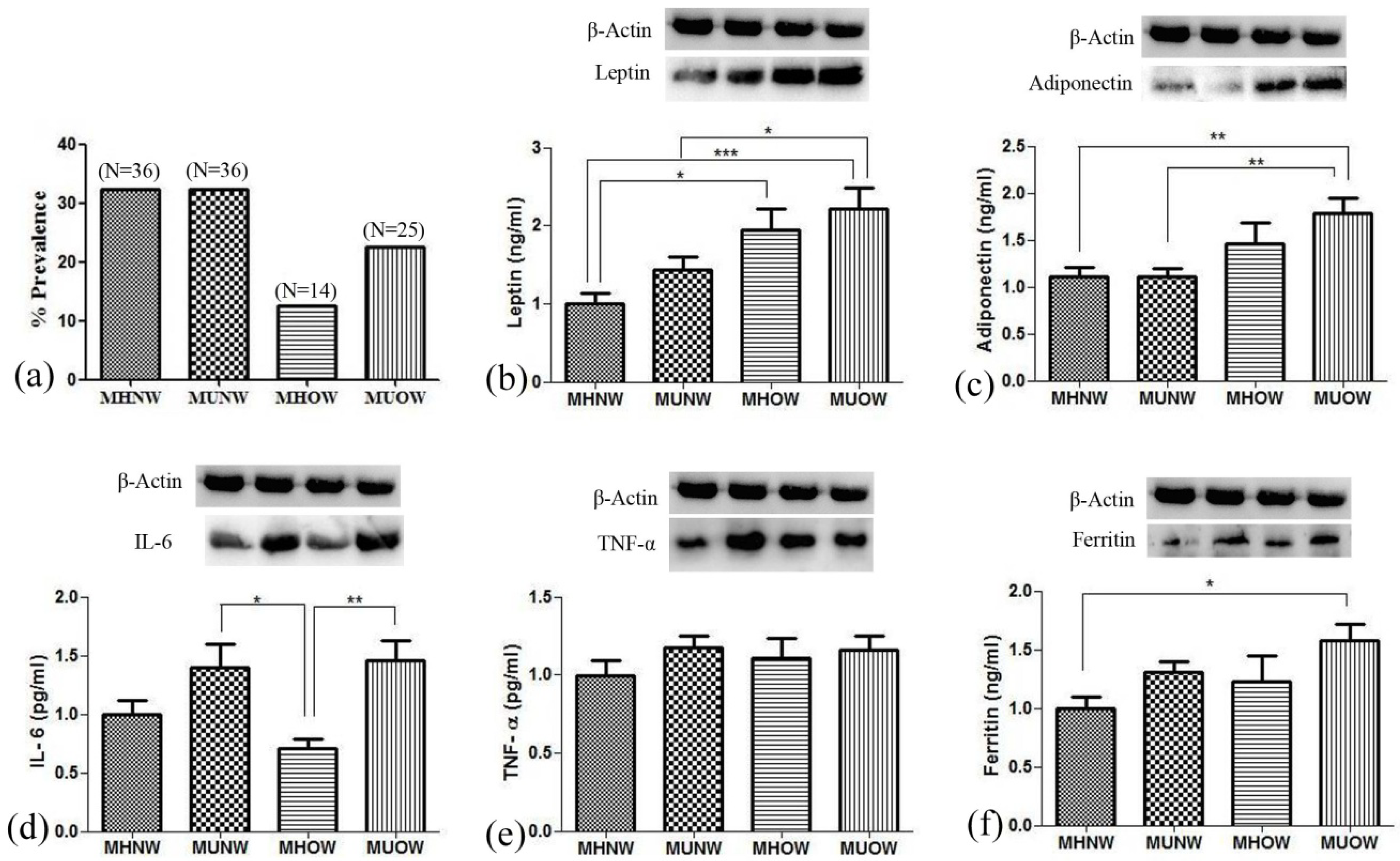
2.5. Analysis of Biochemical Variables
Commercially available kits were used for the analysis of lipid profile, i.e., total cholesterol (Randox lab ltd., County Antrim, UK), triglycerides, high-density lipoprotein-cholesterol (HDL-C), and low-density lipoprotein- cholesterol (LDL-C) (Agappe diagnostics ltd, Kolkata, India). ELISA kits were used for insulin (Sigma Diagnostics Inc., Livonia, MI, USA), leptin (Diagnostics Biochem Canada Inc., London, ON, Canada), adiponectin (Elabscience Biotechnology Co. Ltd., Wuhan, China), C-reactive protein (CRP, Sigma Diagnostics Inc., Livonia, MI, USA), interleukin-6 (IL-6, Diaclone SAS, Besançon, France), tumor necrosis factor-α (TNF-α, Diaclone SAS, Besançon, France), ferritin (Bio-detect, Laguna Hills, CA, USA) as per assay manual of the kits. Insulin resistance was calculated using McAuley’s Index i.e., exp (2.63–0.28 ln insulin [μU/mL]–0.31 ln triglycerides [mM/mL]) [
18].
2.6. Antibodies
The primary antibodies used were primary rabbit polyclonal anti-Leptin (1∶1000, Ob (A-20) sc-842), primary rabbit polyclonal anti-AdipoR2 (1∶1000, (H-44) sc-99184), primary rabbit polyclonal anti-IL-6(1∶1000, (H-183) sc-7920), primary rabbit polyclonal anti-β-actin (1:1000 (N-21) sc-130656) from Santa Cruz Biotechnology, Inc (Heidelberg, Germany). Primary monoclonal mouse anti-ferritin (1∶1000 (E63C02001)), primary rabbit anti-TNF-α (1:500, E18-7014-1) from EnoGene Biotech Co., Ltd., New York, NY, USA. The secondary antibody used was goat anti-rabbit IgG-HRP (sc-2004, 1:25,000) and goat anti-mouse IgG-HRP (sc-2004, 1:25,000) of SantaCruz Biotechnology, Santa Cruz, CA, USA.
2.7. Immunoblot Analysis
Sample containing 30 µg protein was loaded on 10% SDS-PAGE and transferred to PVDF membrane on the semi-dry transfer unit (Bio-Rad laboratories, Hercules, CA, USA). The blots were incubated with primary antibody for the protein of interest for 3 h followed by the addition of IgG-horse radish peroxidase conjugated secondary antibody against the specific primary antibody for 2 h at room temperature. Immunoreactive bands were visualized with a gel documentation instrument (Alliance Q9 Advanced, UVITECH Chemiluminescence Documentation Systems), and densitometric analysis was done using Image J analysis software.
2.8. RNA Extraction
The PAXgen blood RNA tubes containing samples were thawed for two hours at room temperature to ensure the whole cells lysis. Total RNA was isolated using PAXgene Blood RNA Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s guidelines, and extracted RNA was stored at −80 °C.
2.9. Oligonucleotide Microarray Hybridization
The quality and concentration of RNA were checked using a spectrophotometer (NanoDrop2000, Thermo Scientific, Wilmington, DE, USA), and RNA integrity was assessed using Agilent 2100 Bioanalyzer system (Agilent Technologies Inc., Lexington, MA, USA). The RIN values more than 7 were considered adequate for hybridization and scanning. A total of 250 ng of high-quality RNA was used to synthesize complementary DNA followed by an invitro transcription step in which amplification and labeling were done to produce biotin-labeled cRNA according to the MessageAmp II a RNA Amplification kit (Ambion, Inc., Austin, TX, USA) as recommended by Illumina′s sample labeling procedure. The microarray and hybridization were performed on Illumina HumanHT-12 v4 Expression BeadChip (Illumina, Inc., San Diego, CA, USA). The total fluorescence emission from a single spot is collected as a total signal intensity, which is directly proportional to the degree of hybridization [
19].
2.10. Gene Expression Profiling and Gene Ontology(GO)
Differentially expressed genes (DEGs) were identified using Genome Studio™ Gene Expression Module v 1.0 (Illumina Inc., San Diego, CA, USA) and statistically enriched with fold change ≥ 2.0 and
p-value ≤ 0.05. Functional gene ontology and pathway analysis were performed using online software Enrichr [
20].
p-value ≤ 0.05 was adjusted using the hypergeometric distribution. Enriched pathways network was constructed using Cytoscape 3.2.1 [
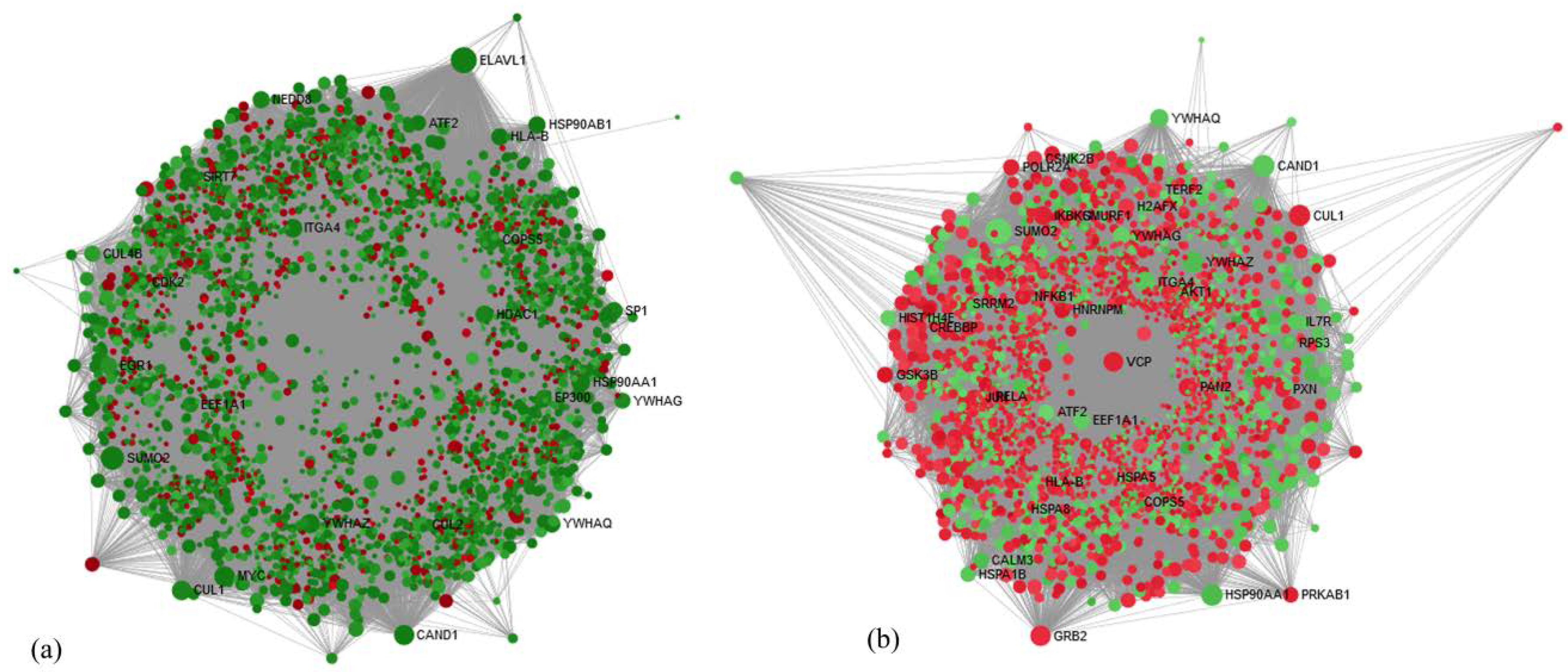
21] with different color coding and shapes for up- and downregulated genes and processes for symbolic representation of the molecular links between genes and processes. The network-based analysis was performed using Network Analyst [
22], which provides visual and statistical analytes for gene expression analysis to obtain highly interconnected hub nodes [
23].
2.11. Functional Gene Set Enrichment Analysis (GSEA) and Upstream Regulation of Shared DEGs
To distinguish the inference of shared DEGs, we executed a functional analysis using the Enrich R platform [
24]. The annotation of the significant gene list was extensively assessed with this software using various libraries, such as Gene ontology library64, biocarta, Kyoto Encyclopedia of Genes and Genomes pathway (KEGG), Wikipathway, Reactome pathway, and Panther. The analysis was based on
p < 0.05 and performed with a Fischer Exact test. Upstream and kinase enrichment analysis were used to prioritize transcription factors and protein kinases by using Expression2Kinase (X2K) bioinformatic tool.
2.12. Validation of Some Candidate Genes Responsible for Obesity by Real-Time-PCR
Among the study population, five samples for each group of MUNW, MHOW, and MUOW with matched age, ethnicity, and occupational routine were selected for validation of the obtained microarray data by reverse transcription-polymerase chain reaction (RT-PCR) for five DEGs. CUL1 (Cullin 1) was selected from hub gene analysis, G6PC3 (Glucose-6-phosphatase catalytic subunit 3) and RHEB (Ras Homolog Enriched In Brain) were selected from the insulin resistance pathway, while STAT3 (Signal Transducer And Activator Of Transcription 3) and Amyloid β A4 precursor protein-binding family B member 1-interacting protein (APBB1IP) were selected from the leptin pathway. Further confirmation of DEGs acquired from microarray was executed using two-step RT-PCR on Biorad CFX96 (Hercules, CA, USA). β-actin was used for normalization of selected candidate DEGs.
2.13. cDNA Synthesis and Quantitative Real-Time Quantitative PCR (qRT-PCR)
cDNA synthesis was performed using a commercially available kit (PrimeScript ™ 1st cDNA Synthesis Kit, Clontech Laboratories, Inc., A Takara Bio company) following the manual provided by the manufacturer. The synthesized cDNA was diluted 10 times for utilization for real-time PCR. Briefly, the 1 µL cDNA was mixed with 0.5 µL TaqMan probe and 5 µL mastermix. The volume of the reaction mixture was made up to 10 µL with Rnase free water. To validate the microarray findings, the expression levels of genes were quantified relative to the endogenous control gene, β-actin (ACTB), using pre-designed TaqMan gene expression assays (Applied Biosystems, Foster City, CA, USA).The mean fold change for each sample was calculated by using the 2DDCt method. The cDNAs were confirmed using 2.5% agarose in agarose gel electrophoresis.
2.14. Statistical Analysis
Statistical analyses were performed using Graph Pad Prism version 5.0 software for Windows (Graph pad Prism software, Laolla, CA, USA) with the level of statistical significance set at p ≤ 0.05. Parameters were expressed as mean ± standard deviation. One wayanalysis of variance (ANOVA) followed by post hoc Bonferroni test was made for comparison between the four groups.
4. Discussion
Although obesity is a disease, not every obese individual is unhealthy or shows metabolic risk markers. Some obese individuals are shielded against the cardiometabolic risk factors, and hence, called metabolically healthy overweight/obese (MHOW/MHO). Similarly, some healthy individuals show metabolic risk markers even though their BMI was within the range of healthy person, and hence, called metabolically unhealthy normal-weight (MUNW) individuals. The mechanisms underlying the advancement of MHOW/MHO and MUNW are still poorly understood. Even though many studies are conducted using omental or subcutaneous adipose tissue to state a variety of pathways involved and affected in MHO and MUNW [
25,
26,
27], the lack of genetic insight using whole blood to study the genetic expression in various pathways majorly in two uncommon obesity, i.e., MUNW and MHOW needs to be explored which makes this study novel itself.
Due to the unavailability of a universally accepted definition of metabolically healthy obesity (MHO), many studies considered 0, 1, or 2 metabolic syndrome components with or without insulin resistance for defining MHO criterion [
6,
28,
29,
30,
31,
32,
33]. Therefore, it had been suggested that MHO is a transition state between MHNW and metabolically unhealthy obesity (MUO) with simply having fewer metabolic abnormalities. Some studies have also suggested that obesity is a biological adaptation leading to changes in adipose tissue biology in response to weight gain [
34,
35]. Considering the zero metabolic syndrome criterion, the present study explores metabolically healthy and unhealthy phenotype. The prevalence of MHOW individuals in our study is comparable to an Indian study published by Geetha etal.2011 [
28]. The haphazard prevalence of metabolically healthy and unhealthy profiles ranges widely, with 10% to 47.7% all over the world [
33,
36,
37,
38,
39,
40]. Such arbitrary results of prevalence may be due to varied country, region, age, gender, race, screening techniques, and absence of a distinct criterion for metabolically healthy and unhealthy individuals [
41].
The development of a pathophysiological state in a body modulates the overall transcriptome, which results in varying genetic expression. Microarray helps in global transcriptome analysis to explore the biological insights into various diseases pathogenesis. We used an end-to-end approach starting from the evaluation of whole blood gene expression profile in MUNW, MHOW, and MUOW with matched controls (MHNW), followed by integrated bioinformatics analysis to extract novel biological genes and pathways and finally, autonomously validated significantly altered common DEGs by real-time PCR. To our surprise, on applying filters of 2-fold change and
p-value ≤0.05, MUOW left with only 12 genes out of 13,671 DEGs; therefore, we continued the further functional analysis with two groups (MUNW and MHOW). Since the volunteers were into regular physical activity, their metabolic state may have switched from MUOW to MHOW, due to which the expression of genes were not in required fold change. On reviewing the literature, we found that a person’s nutritional status can switch between metabolically healthy and metabolically unhealthy status and vice versa [
42,
43]. A 10-year follow-up study of North West Adelaide also reported the transition of metabolically unhealthy obese MUO to MHO [
44]. Thus, it is stated that metabolic health is not a steady-state and can be preserved by targeted interventions [
42].
Interestingly, we got 7470 and 5864 DEGs in MUNW and MHOW, respectively, under the significance threshold of adjusted
p-value ≤ 0.05 and fold change ≥ 2 compared to the healthy control. In MUNW, the gene enrichment analysis showed overexpression of GO-biological pathway ‘JAK-STAT signaling’ in the Panther database. The JAK-STAT signaling pathway is activated by cytokines and interleukins, which further forms STATs dimers and changes the gene expression in the nucleus upon translocation [
45,
46]. The plasma levels of IL-6, TNF-α, and ferritin in the study participants were high in metabolically unhealthy profile as also reported by many studies [
47], suggesting the implication of the JAK-STAT signaling pathway. On the other hand, Biocarta enrichment analysis showed overexpression of ‘The information-processing pathway at the IFN-β enhancer’ pathway, which suggests chromatin remodeling resulting in the activation of transcription, due to interferon-β [
48,
49]. The other enriched pathways analysis revealed common pathways in both the groups (MUNW and MHOW), such as ‘Translation’, ‘Ribosome’, ‘SRP-dependent cotranslational protein targeting to membrane’, ‘Major pathway of rRNA processing in the nucleolus’, ‘rRNA processing’. These outcomes imply that the two groups are varying from each other in some parameters; however, they showed similarity in their gene expression at the translation level as also depicted by GO term enrichment. This suggests that MUNW and MHOW are only biological adaptations to cope up with unhealthy parameters, and they are intermediate states between MHNW and MUO nutritional status.
Next, we analyzed the overexpressed transcription factors via upstream analysis, which showed upregulation of the ‘Aryl hydrocarbon receptor (AHR)’ transcription factor in the MUNW group. AHR plays a crucial role in obesity metabolism by promoting adipogenesis, and it is reported that AHR inhibition leads to reversal of obesity and hepatic steatosis in mice [
50]. In contrast, the upstream analysis showed overexpression of ‘Estrogen receptor 1(ESR1)’ transcription factor in the MHOW group. ESR1 gene polymorphism exhibited a reduced BMI in male and female subjects and also known to attenuate the risk of obesity [
51,
52]. ESR1 forms complex with 17-β estradiol (E2) (E2/ESR1) and modulates adipose tissue vascular endothelial growth factor A (VEGFA), thereby helping in angiogenesis, attenuation of inflammation, and ameliorating adipose tissue function [
53]. Thus, the upregulation of AHR and ESR1 gives us a lead signal to variant metabolic profile in MUNW and MHOW, respectively.
To gain additional genetic insight towards the nutritionally linked significant pathway in MUNW and MHOW, the nutritionally important pathways were selected and analyzed. Two nutritional network pathways that were significantly enriched in MUNW and MHOW subjects were ‘signaling of interleukins’ and ‘metabolic pathways’. Interleukins are produced by leukocytes and target the sites in a paracine and autocrine manner. The oligomerization of interleukins activates various cascades, including the JAK-STAT signaling pathway and MAPK pathway [
54]. The ‘metabolic pathways’ involve enzyme-mediated chemical reactions that lead to catabolism and anabolism in the body. Thus, ‘signaling of interleukins’ portrays the major involvement of inflammatory and anti-inflammatory processes regulating in MUNW as also shown by plasma levels of inflammatory markers in the study participants, while the highly regulated metabolic pathways in MHOW portrays the normal involvement of various regulatory processes. The nutritionally important genes ‘
PIK3R2’ and ‘
PIK3CB’ in the MUNW and MHOW groups, respectively, are a regulatory and catalytic components of Phosphoinositide-3-Kinase (PI3K), a lipase kinase. The regulatory role of
PIK3R2 was reported as ameliorating insulin sensitivity in
PIK3R2 knockout mice [
55], while a promoter variant of
PIK3CB is reported to provide protection from insulin resistance in obese and non-obese individuals [
56]. These results imply the important role of
PIK3R2 and
PIK3CB in regulating insulin sensitivity in obese and non-obese individuals.
The disease pathophysiology can be best understood by analyzing the level of gene expression of the DEGs overlapped between the groups; therefore, we selected genes from hub gene analysis, insulin, and leptin pathways. Among Hub genes, Cullin1 (
CUL1, fold change expression of MHNW = 1.00, MUNW = 0.29, MHOW = 1.56, and MUOW = 0.85) is crucial for cell scaffolding and ubiquitin associated proteolysis [
57]. Moreover,
CUL1 (along with SKP1 (S-phase-kinase-associated protein 1) and F-Box protein) forms the largest E3 ubiquitin ligase family (Skp1-Cullin1-F-box (SCF)) E3 ligase is known for controlling obesity with an association of Skp2 [
58]. It is reported that the mice knockout for SCF Fbxo40 shown elevated levels of IRS1 of the insulin signaling pathway and play a vital role in insulin resistance [
38]. Thus, the overexpression of
CUL1 in MHOW and under-expression in MUNW suggests the diverse nature of MHOW individuals. Among other DEGs, overexpression of the
APBB1IP gene was quantified (fold change expression of MHNW = 1.0, MUNW = 0.32, MHOW = 1.18, and MUOW = 0.74).
APBB1IP is commonly known as Rap1-GTPase, and plays a vital function in diet-induced obesity, insulin, and leptin resistance. Genetic ablation of Rap1-GTPase shields against dietary obesity and imbalance of glucose, insulin, and leptin sensitivity, reduction in inflammation, and ER stress in the hypothalamus [
59]. The overexpression of the
APBB1IP gene in MHOW in comparison to MUNW suggests leptin resistance as also shown by ELISA and Western blotting of leptin. Moreover, it is reported decreased values of McAuley’s index corresponded to increased insulin resistance, which is also shown by this study [
60].
Taking into account insulin pathway and insulin resistance, one gene which gathered our attention was
G6PC3 (fold change expression of MHNW = 1.00, MUNW = 0.31, MHOW = 1.10, MUOW = 0.79). Glucose 6 phosphatase enzymes have three subunits known as
G6PC,
G6PC2, and
G6PC3, together known as G6PC family. Majorly, the GPC family works in association with fasting glucose levels [
61,
62].
G6PC3 is ubiquitously expressed in cells and tissues, and hence, formerly known as “ubiquitously expressed G6Pase catalytic subunit related protein” (UGRP). Its mRNA has been reported to be more abundant than the other paralogous genes of
G6PC [
63,
64].
G6PC3 is well known for its expression in white blood cells, particularly neutrophils, and its deficiency leads to autosomal recessive disease SCN4 (severe congenital neutropenia type 4) [
61,
62,
65]. This subunit of the paralogous gene of G6PC is lesser known for its regulation in blood glucose levels. To date,
G6PC3 known for regulation in neutrophils and deficiency will cause severe congenital neutropenia type 4 (SCN4). However, in our study, in the MUNW group,
G6PC3 has been downregulated, while upregulated in MHOW group suggesting its implication in nutrition-related pathways of insulin resistance.
Summarizing the present study, MHOW is a better metabolic health status with decreased WBC, RBC, thrombocytes, and inflammatory markers than MUNW. The increased blood levels of IL-6, TNF-α, and ferritin proved that inflammation is present in the unhealthy groups. Although ferritin is known as an important biomarker in iron-binding and transferring, it is also rising as an inflammatory marker in obesity [
66]. The tendency and levels of inflammatory markers in all the groups suggest that metabolically healthy individuals have lower inflammation in comparison to metabolically unhealthy individuals. The lower inflammation is also proved at a genetic level where the ‘signaling of interleukins’ pathway was overexpressed in MUNW, while ‘metabolic pathways’ were overexpressed in MHOW. The significant difference in inflammation was observed only in IL-6 concentrations was due to the higher concentration of adiponectin, which acts as an anti-inflammatory protein, hence, limiting the expression and release of pro-inflammatory markers. This, therefore, helps in the development of a better metabolic profile [
67]. Furthermore, the GO-biological pathways and GO terms showed expression of common processes suggesting MUNW and MHOW as the transition state between metabolically healthy and metabolically unhealthy health status.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}