
Development of a 76k Alpaca (Vicugna pacos) Single Nucleotide Polymorphisms (SNPs) Microarray

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Selection of Animals and DNA Sequencing
2.3. Identification and Selection of SNPs
2.4. Performance of the Alpaca SNP Microarray
2.5. Comparison of Genotyping by Sequencing (GBS) and Microarray Genotyping (MG)
2.6. Animal Sample Population Structure
3. Results
3.1. Sequencing of Reduced Representation Libraries
3.2. Selection of SNPs for the Microarray
3.3. Construction of the Alpaca SNPs Microarray
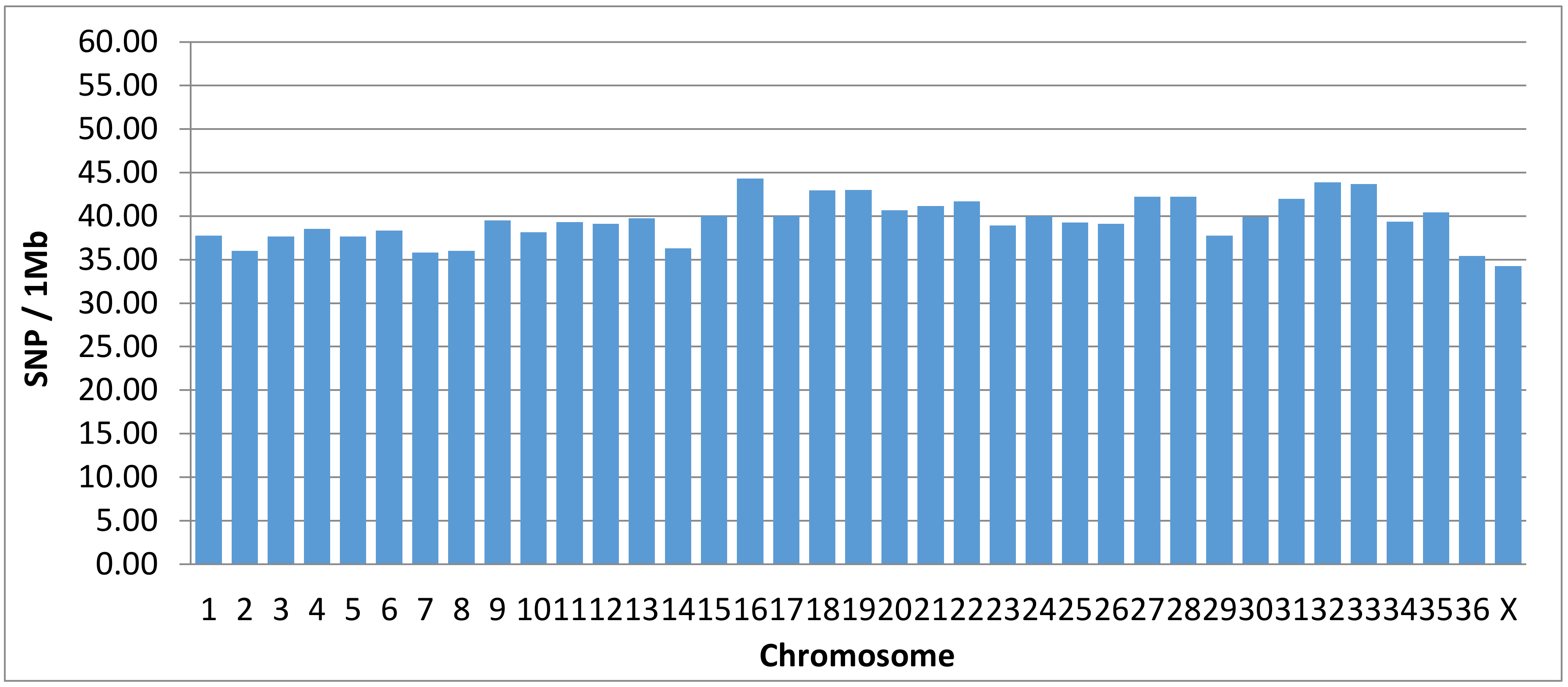
3.4. Performance of the Alpaca SNP Microarray
3.4.1. Concordance between Pedigree and Microarray Genotyping for Trios
3.4.2. Comparison between GBS and Microarray Genotyping
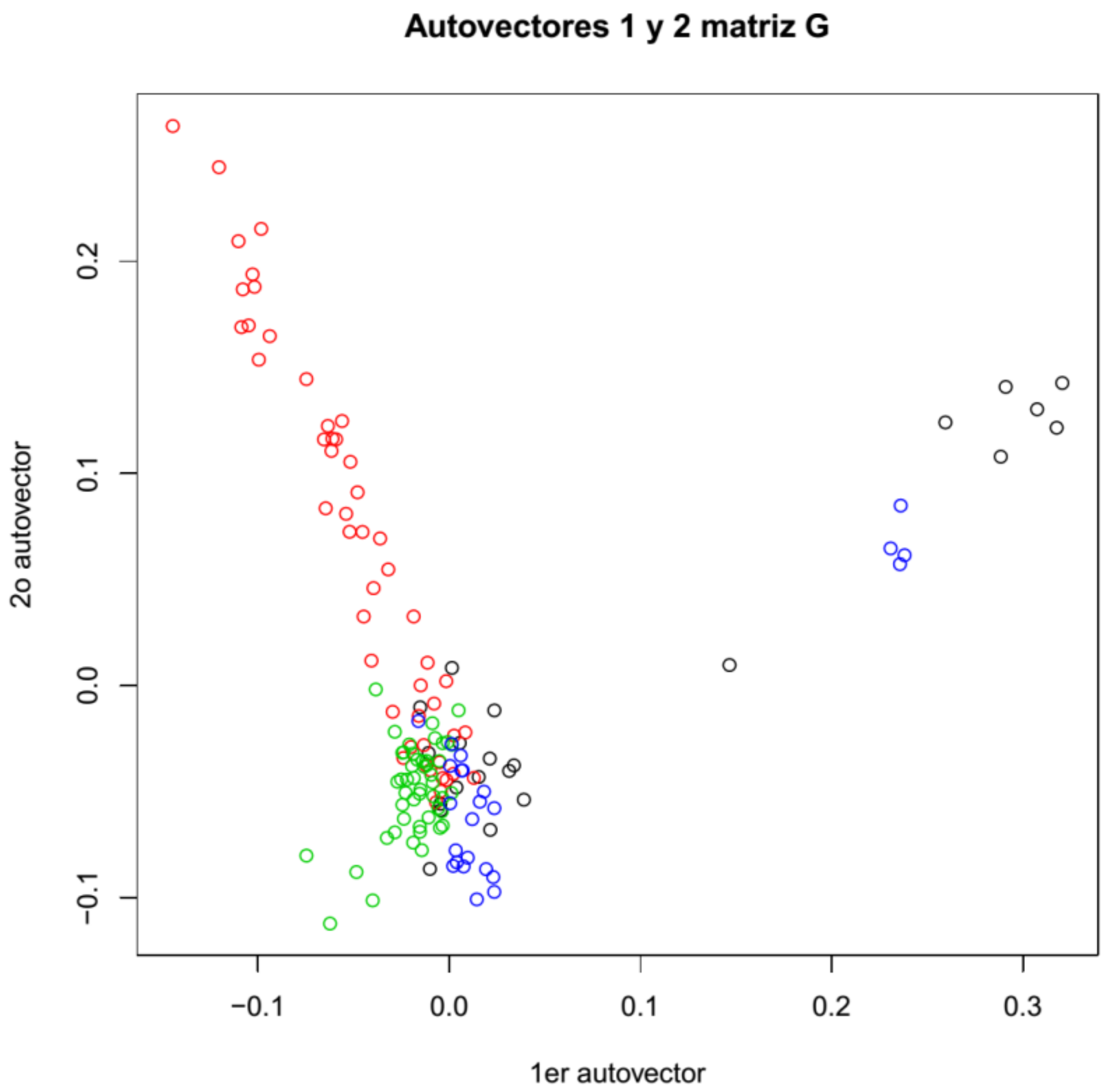
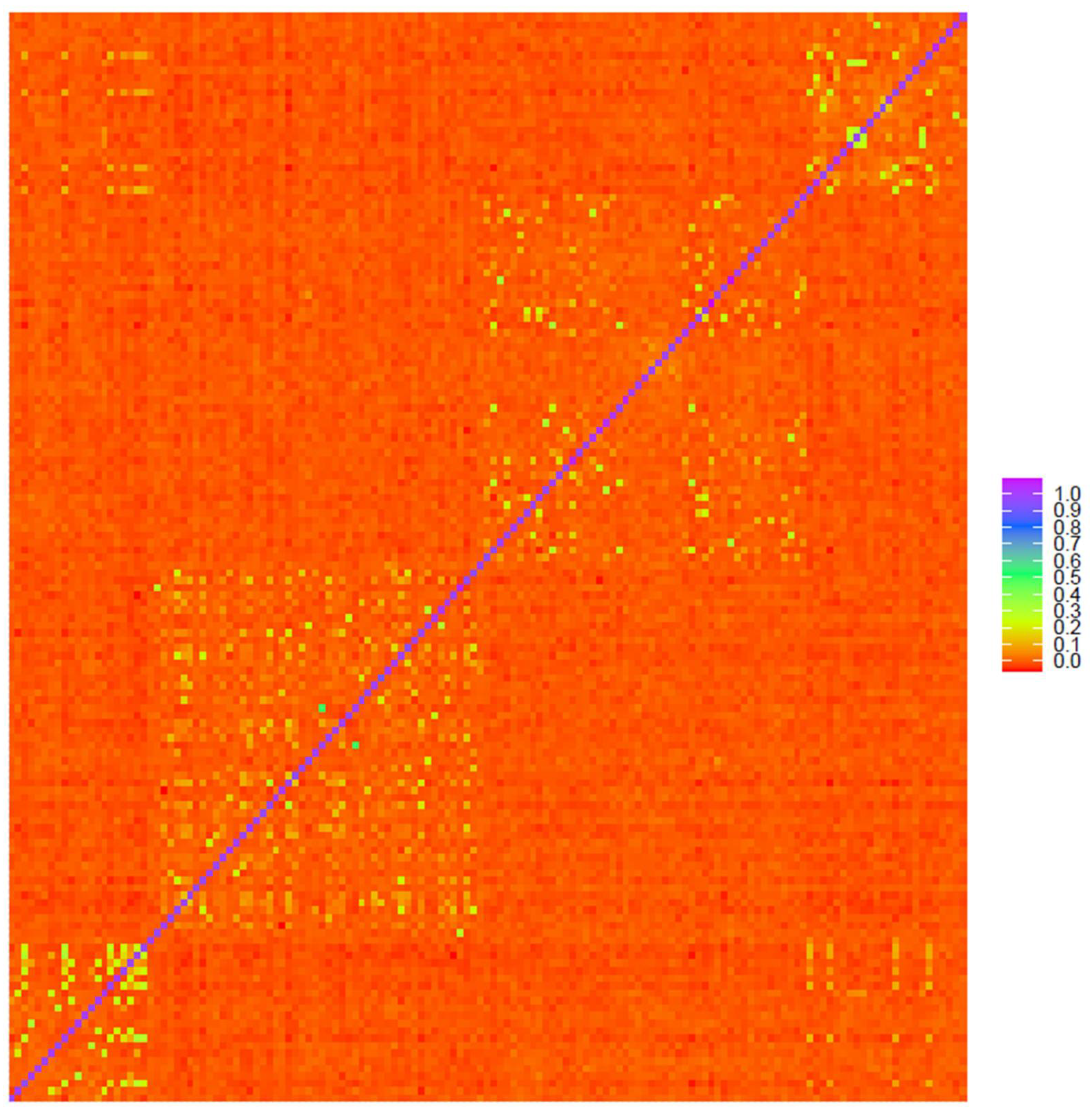
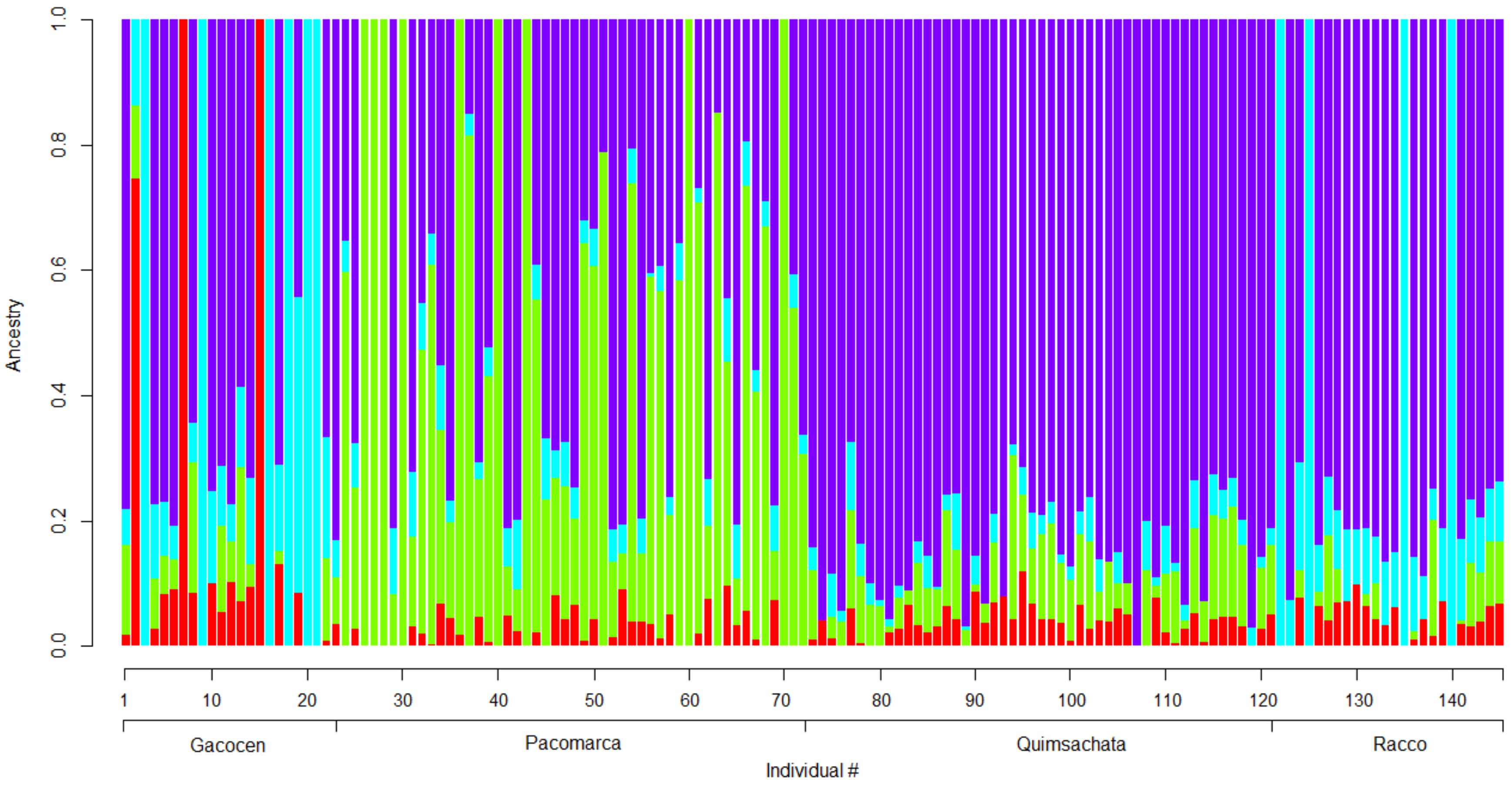
3.5. Sample Population Structure
4. Discussion
4.1. Animal Samples
4.2. Selection of SNPs for the Microarray
4.3. Performance of the Alpaca SNP Microarray
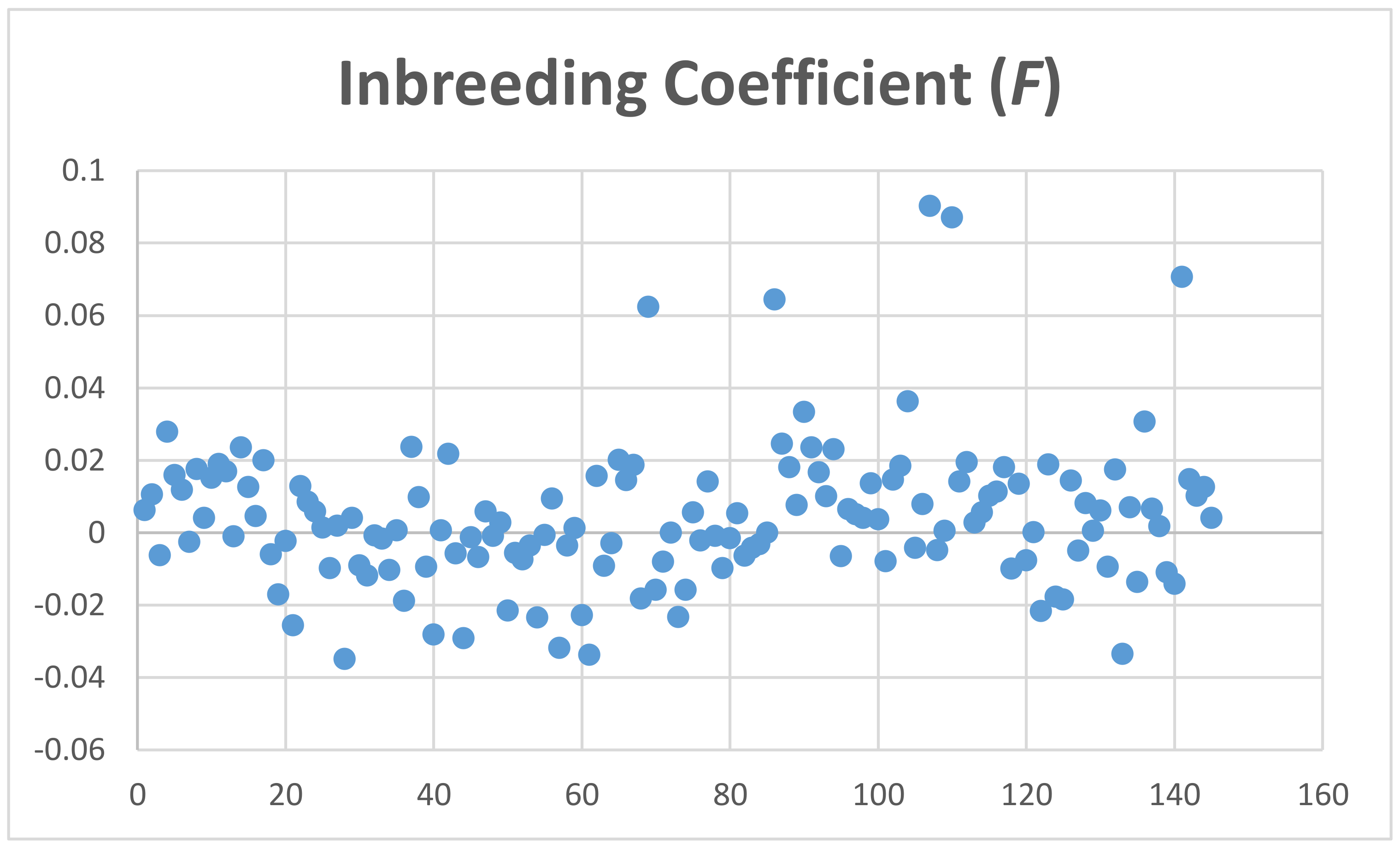
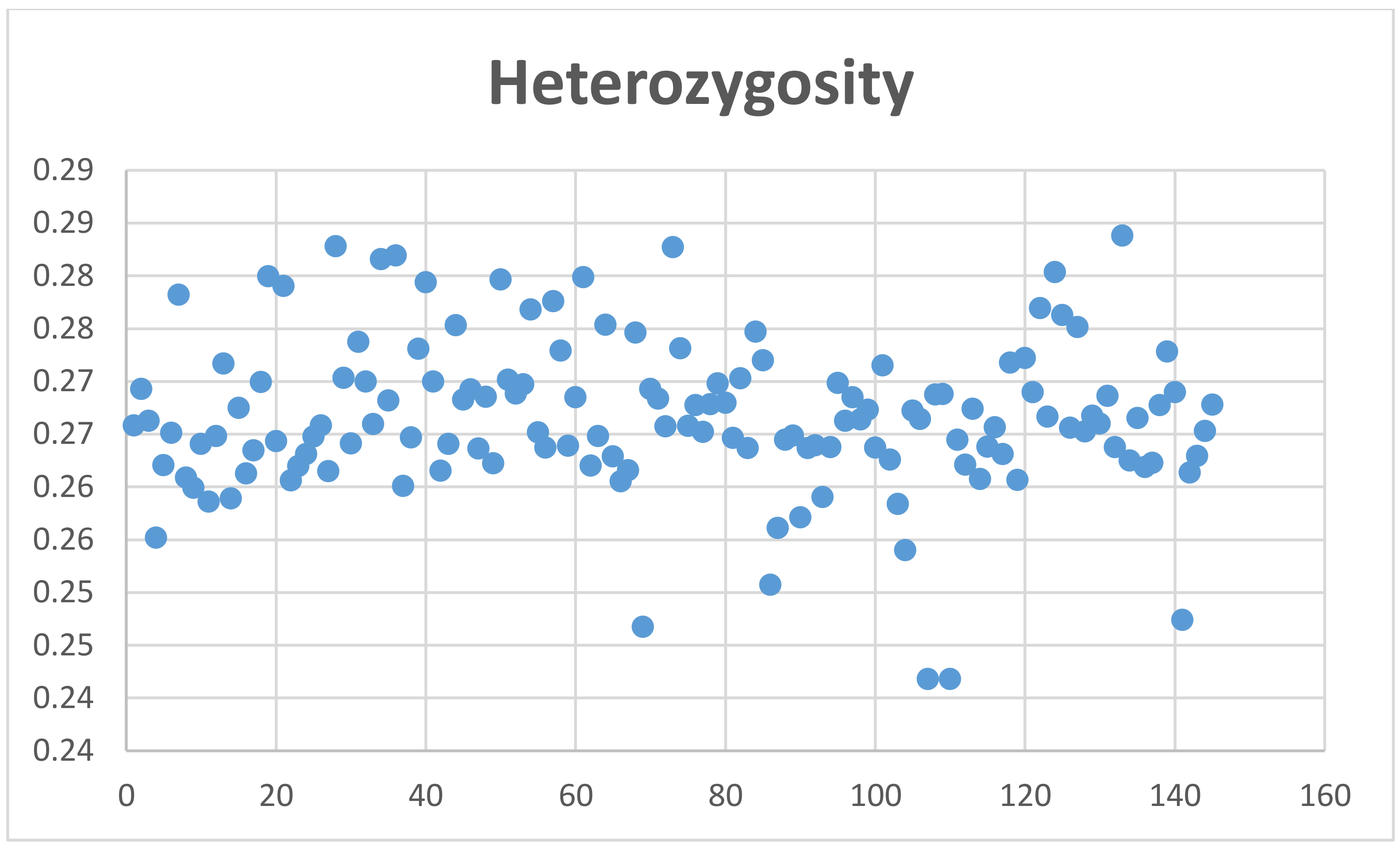
4.4. Sample Population Structure
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- CENAGRO. Censo Nacional Agropecuario 2012. In CENAGRO Resultados Definitivos: IV Censo Nacional Agropecuario-2012; Instituto Nacional de Estadística e Informática (INEI): Julio, Perú, 2013; Volume 63, p. 18. Available online: https://sinia.minam.gob.pe/documentos/resultados-definitivos-iv-censo-nacional-agropecuario-2012-0 (accessed on 13 November 2020).
- Quispe, E.C.; Rodríguez, T.C.; Iñiguez, L.R.; Mueller, J.P. Producción de fibra de alpaca, llama, vicuña y guanaco en Sudamérica. Anim. Genet. Resour. Inf. 2009, 45, 1–14. [Google Scholar] [CrossRef]
- Ordoñez, C.; Cucho, H.; Ampuero, E.; Antezana, W.; Cayo, S. Inseminación Artificial de Alpacas con Semen Fresco, Refrigerado y Descongelado Colectado por Electroeyaculación. Spermova 2013, 3, 65–66. [Google Scholar]
- Huanca, W. Reproductive biotechnologies in domestic South American camelids as alternatives for genetic improvement. Arch. Latinoam. Prod. Anim. 2015, 23, 1–4. [Google Scholar]
- Richardson, M.F.; Munyard, K.; Croft, L.J.; Allnutt, T.R.; Jackling, F.; Alshanbari, F.; Jevit, M.; Wright, G.A.; Cransberg, R.; Tibary, A.; et al. Chromosome-Level Alpaca Reference Genome VicPac3.1 Improves Genomic Insight Into the Biology of New World Camelids. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Balmus, G.; Trifonov, V.A.; Biltueva, L.S.; O’Brien, P.C.M.; Alkalaeva, E.S.; Fu, B.; Skidmore, J.A.; Allen, T.; Graphodatsky, A.S.; Yang, F.; et al. Cross-species chromosome painting among camel, cattle, pig and human: Further insights into the putative Cetartiodactyla ancestral karyotype. Chromosom. Res. 2007, 15. [Google Scholar] [CrossRef]
- Avila, F.; Baily, M.P.; Perelman, P.; Das, P.J.; Pontius, J.; Chowdhary, R.; Owens, E.; Johnson, W.E.; Merriwether, D.A.; Raudsepp, T. A comprehensive whole-genome integrated cytogenetic map for the alpaca (Lama pacos). Cytogenet. Genome Res. 2014, 144, 196–207. [Google Scholar] [CrossRef]
- Mendoza, M.N.; Raudsepp, T.; Alshanbari, F.; Gutiérrez, G.; Ponce de León, F.A. Chromosomal Localization of Candidate Genes for Fiber Growth and Color in Alpaca (Vicugna pacos). Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Mendoza, M.N.; Raudsepp, T.; More, M.J.; Gutiérrez, G.A.; Ponce de León, F.A. Cytogenetic Mapping of 35 New Markers in the Alpaca (Vicugna pacos). Genes (Basel). 2020, 11, 522. [Google Scholar] [CrossRef]
- More, M.; Gutiérrez, G.; Rothschild, M.; Bertolini, F.; Ponce de León, F.A. Evaluation of SNP Genotyping in Alpacas Using the Bovine HD Genotyping Beadchip. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Guridi, M.; Soret, B.; Alfonso, L.; Arana, A. Single nucleotide polymorphisms in the Melanocortin 1 Receptor gene are linked with lightness of fibre colour in Peruvian Alpaca (Vicugna pacos). Anim. Genet. 2011, 42. [Google Scholar] [CrossRef]
- Chandramohan, B.; Renieri, C.; La Manna, V.; La Terza, A. The alpaca agouti gene: Genomic locus, transcripts and causative mutations of eumelanic and pheomelanic coat color. Gene 2013, 521. [Google Scholar] [CrossRef]
- Chandramohan, B.; Renieri, C.; La Manna, V.; La Terza, A. The Alpaca Melanocortin 1 Receptor: Gene Mutations, Transcripts, and Relative Levels of Expression in Ventral Skin Biopsies. Sci. World J. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Feeley, N.L.; Bottomley, S.; Munyard, K.A. Novel mutations in Vicugna pacos (alpaca) Tyrp1 are not correlated with brown fibre colour phenotypes. Small Rumin. Res. 2016, 143. [Google Scholar] [CrossRef]
- Fernández Suárez, A.G.; Gutiérrez Reynoso, G.A.; Ponce de León Bravo, F.A. Identificación bioinformática de Polimorfismos de Nucleótido Simple (PNSs) en genes candidatos para las características de la fibra en alpacas (Vicugna pacos). Rev. Peru. Biol. 2019, 26. [Google Scholar] [CrossRef]
- Jones, M.; Sergeant, C.; Richardson, M.; Groth, D.; Brooks, S.; Munyard, K. A non-synonymous SNP in exon 3 of the KIT gene is responsible for the classic grey phenotype in alpacas (Vicugna pacos). Anim. Genet. 2019, 50. [Google Scholar] [CrossRef]
- Matukumalli, L.K.; Lawley, C.T.; Schnabel, R.D.; Taylor, J.F.; Allan, M.F.; Heaton, M.P.; O’Connell, J.; Moore, S.S.; Smith, T.P.L.; Sonstegard, T.S.; et al. Development and Characterization of a High Density SNP Genotyping Assay for Cattle. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [Green Version]
- Ramos, A.M.; Crooijmans, R.P.M.A.; Affara, N.A.; Amaral, A.J.; Archibald, A.L.; Beever, J.E.; Bendixen, C.; Churcher, C.; Clark, R.; Dehais, P.; et al. Design of a High Density SNP Genotyping Assay in the Pig Using SNPs Identified and Characterized by Next Generation Sequencing Technology. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [Green Version]
- Groenen, M.A.; Megens, H.-J.; Zare, Y.; Warren, W.C.; Hillier, L.W.; Crooijmans, R.P.; Vereijken, A.; Okimoto, R.; Muir, W.M.; Cheng, H.H. The development and characterization of a 60K SNP chip for chicken. BMC Genomics 2011, 12. [Google Scholar] [CrossRef] [Green Version]
- Darrier, B.; Russell, J.; Milner, S.G.; Hedley, P.E.; Shaw, P.D.; Macaulay, M.; Ramsay, L.D.; Halpin, C.; Mascher, M.; Fleury, D.L.; et al. A Comparison of Mainstream Genotyping Platforms for the Evaluation and Use of Barley Genetic Resources. Front. Plant Sci. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Fan, R.; Gu, Z.; Guang, X.; Marín, J.C.; Varas, V.; González, B.A.; Wheeler, J.C.; Hu, Y.; Li, E.; Sun, X.; et al. Genomic analysis of the domestication and post-Spanish conquest evolution of the llama and alpaca. Genome Biol. 2020, 21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26. [Google Scholar] [CrossRef] [Green Version]
- Picard Toolkit. Broad Institute, GitHub Repository. 2019. Available online: http://broadinstitute.github.io/picard/ (accessed on 18 January 2019).
- BCFtools. Doc. 2019. Available online: http//samtools.github.io/bcftools (accessed on 18 February 2019).
- R Core Team. 2020 R: A Language and Environment for Statistical Computing. Available online: http//www.R-project.org/ (accessed on 18 October 2020).
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-wide Complex Trait Analysis. Am. J. Hum. Genet. 2011, 88. [Google Scholar] [CrossRef] [Green Version]
- Patterson, N.; Price, A.L.; Reich, D. Population Structure and Eigenanalysis. PLoS Genet. 2006, 2. [Google Scholar] [CrossRef]
- Deniskova, T.; Dotsev, A.; Lushihina, E.; Shakhin, A.; Kunz, E.; Medugorac, I.; Reyer, H.; Wimmers, K.; Khayatzadeh, N.; Sölkner, J.; et al. Population Structure and Genetic Diversity of Sheep Breeds in the Kyrgyzstan. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- RStudio Team. 2020 RStudio: Integrated Development for R. Available online: http://www.rstudio.com/ (accessed on 18 August 2020).
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Skotte, L.; Korneliussen, T.S.; Albrechtsen, A. Estimating Individual Admixture Proportions from Next Generation Sequencing Data. Genetics 2013, 195, 693. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.H.; Shringarpure, S.S.; Novembre, J.; Lange, K. Admixture 1.3 Software Manual 2020. Available online: http://dalexander.github.io/admixture/admixture-manual.pdf (accessed on 13 November 2020).
- Fernández Baca, S. Situación actual de los camélidos sudamericanos en Perú. In Proyecto de Cooperación Técnica en Apoyo de la Crianza y Aprovechamiento de los Camélidos Sudamericanos en la Región Andina TCP/RLA/2914; Organización de las Naciones Unidas para la Agricultura y la Alimentación (FAO): Julio, Perú, 2005; Volume 63. [Google Scholar]
- Feeley, N.L.; Munyard, K.A. Characterisation of the melanocortin-1 receptor genein alpaca and identification of possible markers associatedwith phenotypic variations in colour. Anim. Prod. Sci. 2009, 41, 675–681. [Google Scholar] [CrossRef]
- Allain, D.; Renieri, C. Genetics of fibre production and fleece characteristics in small ruminants, Angora rabbit and South American camelids. Animal 2010, 4, 1472–1481. [Google Scholar] [CrossRef] [Green Version]
- Oliehoek, P.A.; Bijma, P. Effects of pedigree errors on the efficiency of conservation decisions. Genet. Sel. Evol. 2009, 41. [Google Scholar] [CrossRef] [Green Version]
- Hartl, D.L.; Clark, A.G. Principles of Population Genetics, 4th ed.; Sinauer: Sunderland, MA, USA, 1997; ISBN 9780878933082. [Google Scholar]
- Slate, J.; David, P.; Dodds, K.G.; Veenvliet, B.A.; Glass, B.C.; Broad, T.E.M. Understanding the relationship between the inbreeding coefficient and multilocus heterozygosity: Theoretical expectations and empirical data. Heredity (Edinb). 2004, 93, 255–265. [Google Scholar] [CrossRef]
- Houle, D. Allozyme associated heterosis in Drosophila melanogaster. Genetics 1989, 123, 789–801. [Google Scholar] [CrossRef]
- Savolainen, O.; Hedrick, P. Heterozygosity and fitness: No association in Scots pine. Genetics 1995, 140, 755–766. [Google Scholar] [CrossRef]
- Whitlock, M. Lack of correlation between heterozygosity and fitness in forked fungus beetles. Heredity (Edinb). 1993, 70, 574–581. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Number of Animals | |
|---|---|---|
| Pasco | San Pedro de Racco | 25 |
| GACOCEN | 25 | |
| Puno | Pacomarca | 50 |
| INIA-Quimsachata | 50 | |
| Total alpacas | 150 | |
| Round | Phred Score | Genotyping Rate (GR) | Minor Allele Frequency (MAF) | Illumina Score | Length of Flanking Sequences | First Set Number of SNPs | Second Set Number of SNPs |
|---|---|---|---|---|---|---|---|
| 1 | >10 | ≥0.45 | 0.05–0.50 | ≥0.60 | 40 | 45,156 | 17,148 |
| 2 | >10 | ≥0.45 | 0.05–0.50 | ≥0.60 | 35 | 1319 | 1876 |
| 3 | >10 | ≥0.15 | 0.04–0.50 | ≥0.60 | 40 | 4027 | 6734 |
| 4 | >10 | ≥0.15 | 0.04–0.50 | ≥0.60 | 35 | 320 | 628 |
| 5 | >10 | ≥0.15 | 0.01–0.039 | ≥0.60 | 40 | 829 | 1821 |
| 6 | >10 | ≥0.15 | 0.01–0.039 | ≥0.60 | 35 | 121 | 222 |
| Total | 51,772 | 28,429 |
| Fragment Lengths in kbp | Number of Fragments Containing One SNP Identified in This Study | Number of Fragments with One SNP Included in The Microarray |
|---|---|---|
| ≥700–800 | 1 | 1 |
| ≥600–700 | 0 | 1 |
| ≥500–600 | 6 | 5 |
| ≥400–500 | 3 | 3 |
| ≥300–400 | 10 | 10 |
| ≥200–300 | 29 | 35 |
| ≥100–200 | 315 | 375 |
| ≥90–100 | 210 | 243 |
| ≥80–90 | 302 | 366 |
| ≥70–80 | 541 | 696 |
| ≥60–70 | 1075 | 1285 |
| ≥50–60 | 2770 | 3011 |
| ≥40–50 | 5999 | 6682 |
| ≥30–40 | 10,848 | 11,069 |
| ≥20–30 | 21,145 | 19,146 |
| ≥10–20 | 32,282 | 29,070 |
| ≥0–10 | 4683 | 4510 |
| Total | 80,201 | 76,508 |
| Description of SNPs | Nº SNPs Selected in This Study | Final Nº SNPs Selected by Affymetrix |
|---|---|---|
| First set | 51,772 | 49,282 |
| Second set | 28,429 | 26,924 |
| Candidate genes | 302 | 302 |
| Controls | 100 | |
| Duplicate controls | 302 | |
| Total SNPs | 80,503 | 76,910 |
| Scaffolds | Number of SNPs | Number of 40 kbp Fragments | Average Interval between SNPs | Length Covered by SNPs (bp) | VicPac3.1 (bp) Length | % Length of Genome Covered with SNPs (VicPac3.1) |
|---|---|---|---|---|---|---|
| Localized on Chromosomes | 59,297 | 38,165 | 26,992 | 1,525,673,735 | 1,602,467,523 | 95.21 |
| Unassigned | 17,211 | 12,491 | 25,160 | 393,461,101 | 517,133,374 | 76.09 |
| Total | 76,508 | 50,656 | 26,580 | 1,919,134,836 | 2,119,600,897 | 90.54 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calderon, M.; More, M.J.; Gutierrez, G.A.; Ponce de León, F.A. Development of a 76k Alpaca (Vicugna pacos) Single Nucleotide Polymorphisms (SNPs) Microarray. Genes 2021, 12, 291. https://doi.org/10.3390/genes12020291
Calderon M, More MJ, Gutierrez GA, Ponce de León FA. Development of a 76k Alpaca (Vicugna pacos) Single Nucleotide Polymorphisms (SNPs) Microarray. Genes. 2021; 12(2):291. https://doi.org/10.3390/genes12020291
Chicago/Turabian StyleCalderon, Marcos, Manuel J. More, Gustavo A. Gutierrez, and Federico Abel Ponce de León. 2021. "Development of a 76k Alpaca (Vicugna pacos) Single Nucleotide Polymorphisms (SNPs) Microarray" Genes 12, no. 2: 291. https://doi.org/10.3390/genes12020291
APA StyleCalderon, M., More, M. J., Gutierrez, G. A., & Ponce de León, F. A. (2021). Development of a 76k Alpaca (Vicugna pacos) Single Nucleotide Polymorphisms (SNPs) Microarray. Genes, 12(2), 291. https://doi.org/10.3390/genes12020291