
A Workflow for Selection of Single Nucleotide Polymorphic Markers for Studying of Genetics of Ischemic Stroke Outcomes

 ,
,  , ,
, , 
Abstract
:1. Introduction
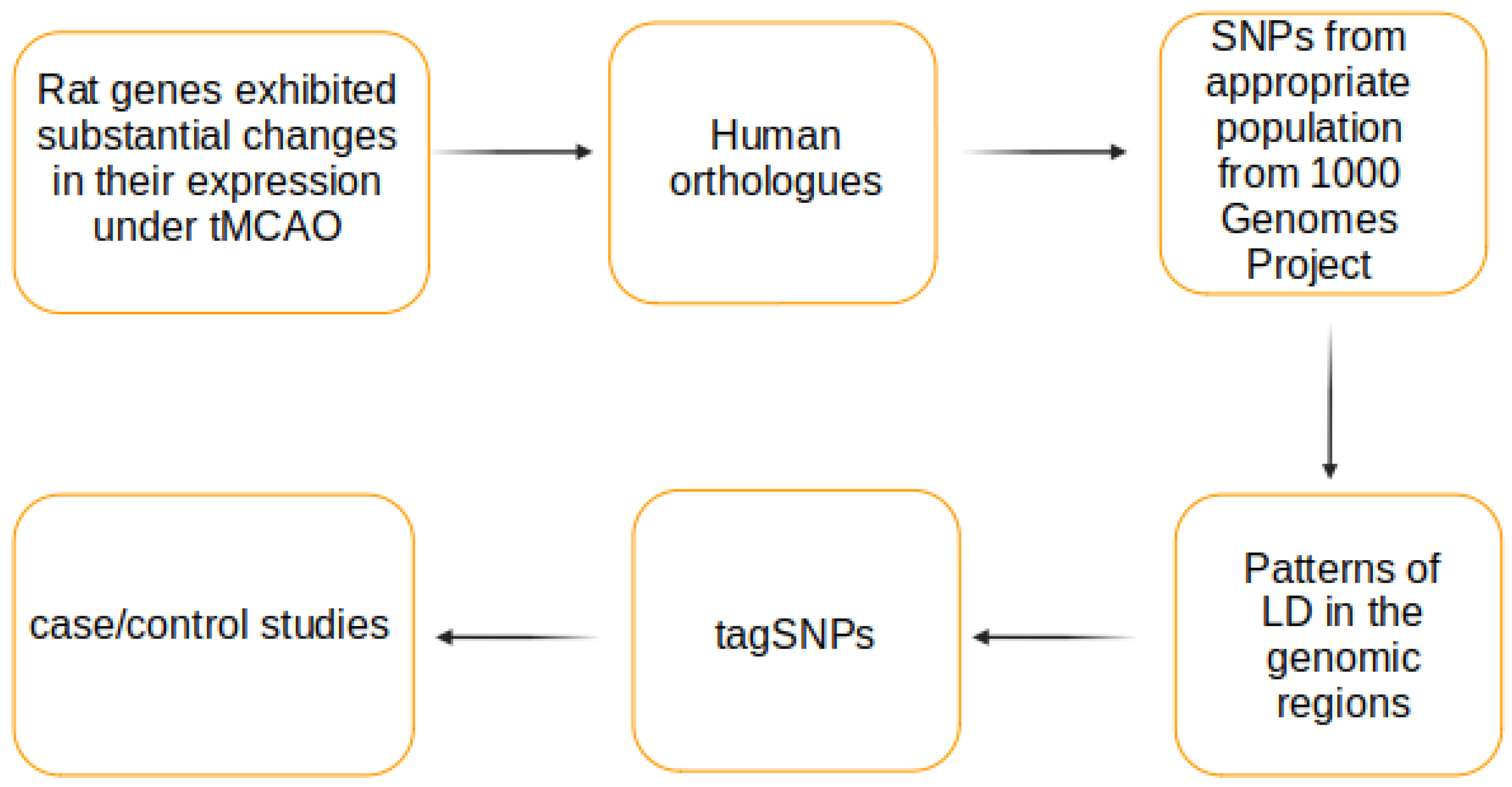
2. Materials and Methods
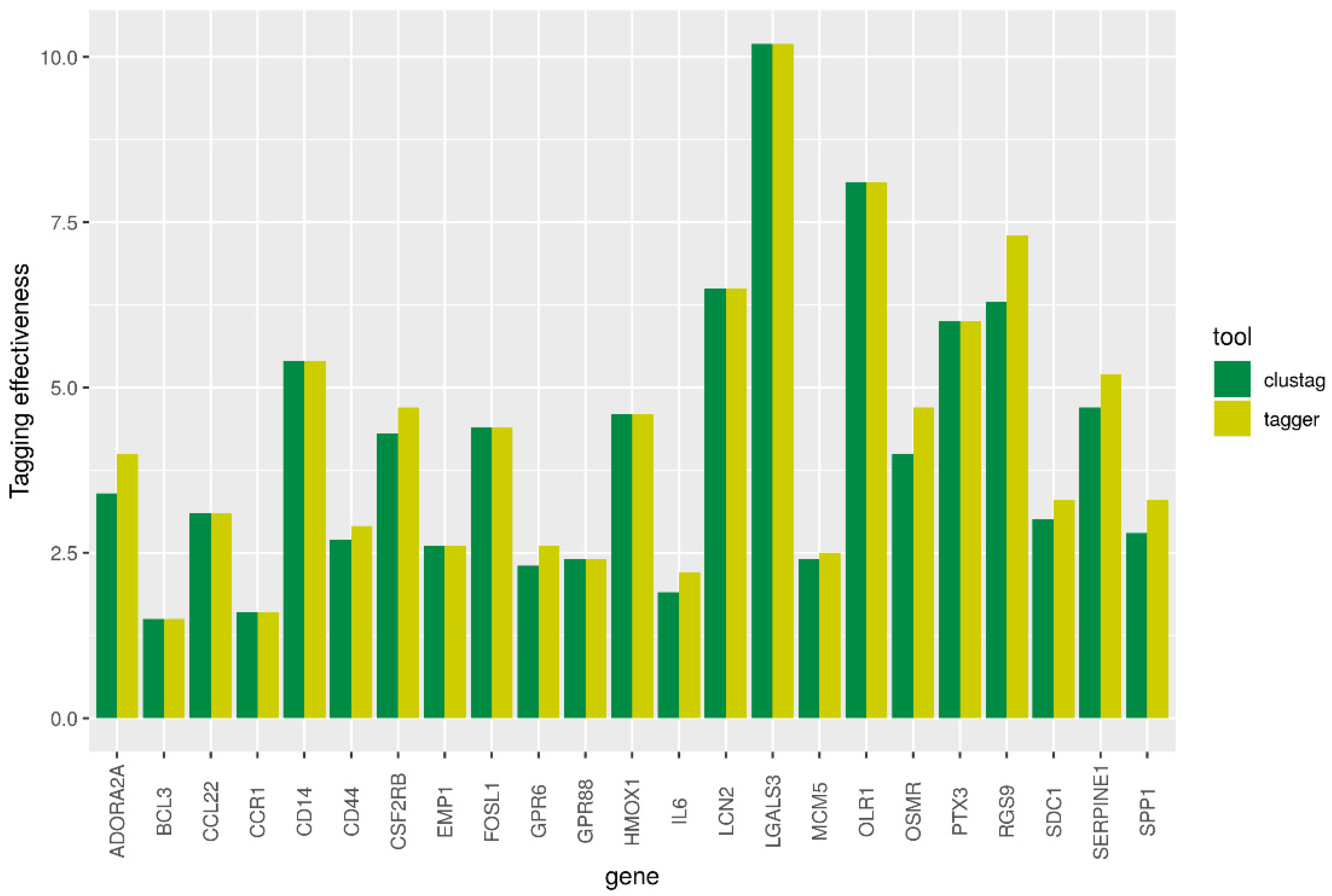
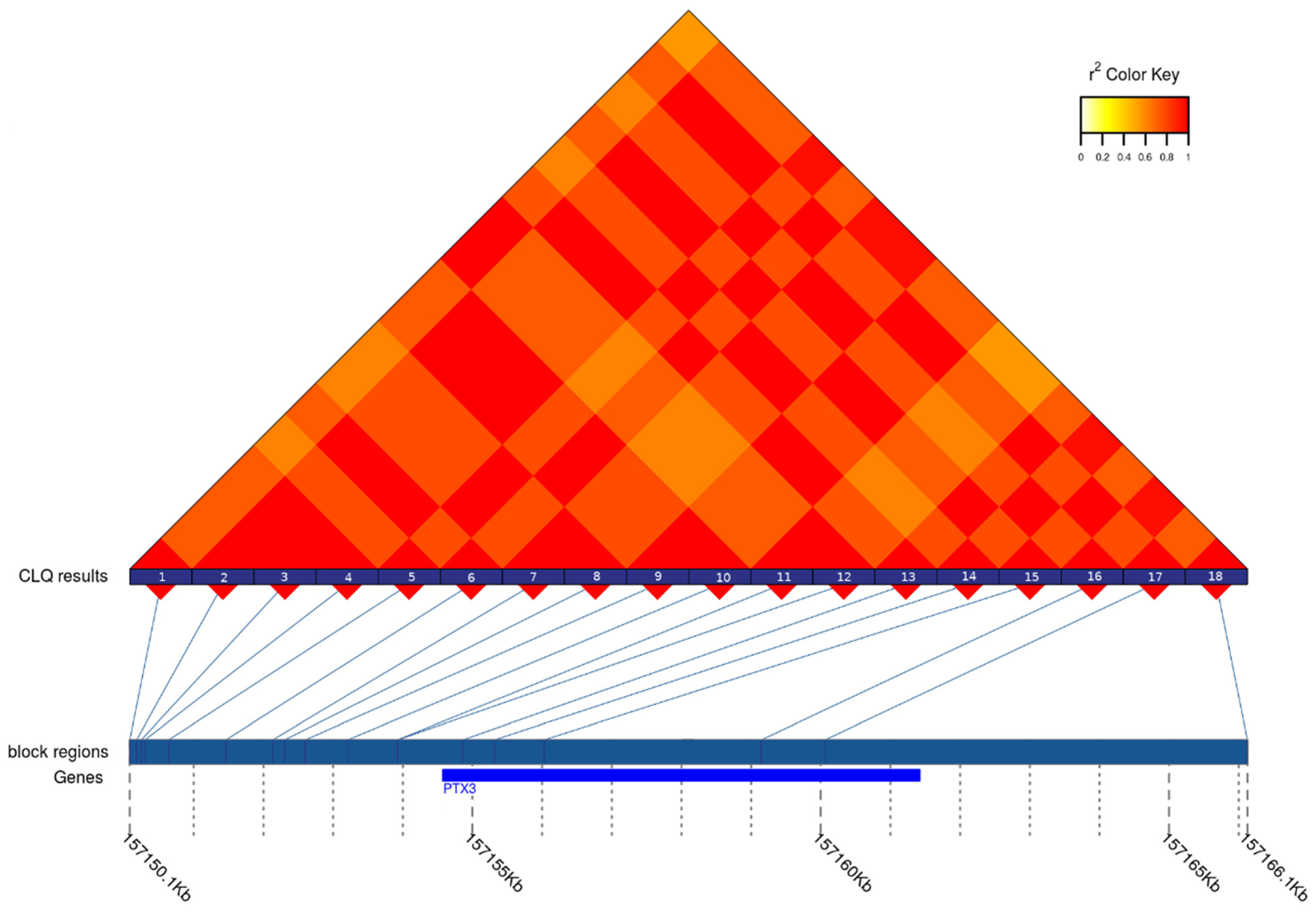
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Jood, K.; Ladenvall, C.; Rosengren, A.; Blomstrand, C.; Jern, C. Family history in ischemic stroke before 70 years of age: The Sahlgrenska academy study on ischemic stroke. Stroke 2005, 36, 1383–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jickling, G.C.; Kittner, S.J. A SNP-it of stroke outcome. Neurology 2019, 92, 549–550. [Google Scholar] [CrossRef]
- Torres-Aguila, N.P.; Carrera, C.; Muiño, E.; Cullell, N.; Cárcel-Márquez, J.; Gallego-Fabrega, C.; González-Sánchez, J.; Bustamante, A.; Delgado, P.; Ibañez, L.; et al. Clinical variables and genetic risk factors associated with the acute outcome of ischemic stroke: A systematic review. J. Stroke 2019, 21, 276–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, M.; Zhao, S. Candidate gene identification approach: Progress and challenges. Intl. J. Biol. Sci. 2007, 3, 420–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcone, G.J.; Malik, R.; Dichgans, M.; Rosand, J. Current concepts and clinical applications of stroke genetics. Lancet Neurol. 2014, 13, 405–418. [Google Scholar] [CrossRef]
- Söderholm, M.; Pedersen, A.; Lorentzen, E.; Stanne, T.M.; Bevan, S.; Olsson, M.; Cole, J.W.; Fernandez-Cadenas, I.; Hankey, G.J.; Jimenez-Conde, J.; et al. Genome-wide association meta-analysis of functional outcome after ischemic stroke. Neurology 2019, 92, E1271–E1283. [Google Scholar] [CrossRef] [Green Version]
- Ibanez, L.; Heitsch, L.; Carrera, C.; Farias, F.H.; Dhar, R.; Budde, J.; Cruchaga, C. Multi-ancestry genetic study in 5,876 patients identifies an association between excitotoxic genes and early outcomes after acute ischemic stroke. medRxiv Prepr Serv Heal. Sci. 2020. [Google Scholar] [CrossRef]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Amer. J.Human Gen. 2017, 101, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.-J.; Li, X.; Zhao, S.-H. Digital candidate gene approach (DigiCGA) for identification of cancer genes. Methods Mol. Biol. 2010, 653, 105–129. [Google Scholar] [CrossRef]
- White, B.C.; Sullivan, J.M.; DeGracia, D.J.; O’Neil, B.J.; Neumar, R.W.; Grossman, L.I.; Rafols, J.A.; Krause, G.S. Brain ischemia and reperfusion: Molecular mechanisms of neuronal injury. J. Neurol. Sci. 2000, 179, 1–33. [Google Scholar] [CrossRef]
- Huang, H.; Winter, E.E.; Wang, H.; Weinstock, K.G.; Xing, H.; Goodstadt, L.; Stenson, P.D.; Cooper, D.N.; Smith, D.; Albà, M.M.; et al. Evolutionary Conservation and Selection of Human Disease Gene Orthologs in the Rat and Mouse Genomes. 2004. Available online: http://genomebiology.com/2004/5/7/R47 (accessed on 21 October 2019).
- Howells, D.W.; Porritt, M.J.; Rewell, S.S.J.; O’Collins, V.; Sena, E.S.; Van Der Worp, H.B.; Traystman, R.J.; MacLeod, M.R. Different strokes for different folks: The rich diversity of animal models of focal cerebral ischemia. J. Cereb. Blood Flow Metabolism. 2010, 30, 1412–1431. [Google Scholar] [CrossRef] [Green Version]
- Dergunova, L.V.; Filippenkov, I.B.; Stavchansky, V.V.; Denisova, A.E.; Yuzhakov, V.V.; Mozerov, S.A.; Limborska, S.A. Genome-wide transcriptome analysis using RNA-Seq reveals a large number of differentially expressed genes in a transient MCAO rat model. BMC Genom. 2018, 19, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, S.E.; McLaren, W.; Gil, L.; Thormann, A.; Schuilenburg, H.; Sheppard, D.; Parton, A.; Armean, I.M.; Trevanion, S.J.; Flicek, P.; et al. Ensembl variation resources. Database 2018, 2018, 1–12. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Capella-Gutiérrez, S.; Pryszcz, L.P.; Marcet-Houben, M.; Gabaldón, T. PhylomeDB v4: Zooming into the plurality of evolutionary histories of a genome. Nucleic Acids Res. 2014, 42, 897–902. [Google Scholar] [CrossRef] [Green Version]
- Pryszcz, L.P.; Huerta-Cepas, J.; Gabaldón, T. MetaPhOrs: Orthology and paralogy predictions from multiple phylogenetic evidence using a consistency-based confidence score. Nucleic Acids Res. 2011, 39, e32. [Google Scholar] [CrossRef] [Green Version]
- BioMart. Available online: http://grch37.ensembl.org/biomart/martview/a117c9fd556f996c278019dae08cfa00 (accessed on 5 July 2019).
- Wojcik, G.L.; Fuchsberger, C.; Taliun, D.; Welch, R.; Martin, A.R.; Shringarpure, S.; Carlson, C.S.; Abecasis, G.; Kang, H.M.; Boehnke, M.; et al. Imputation-aware tag SNP selection to improve power for large-scale, multi-ethnic association studies. G3 Genes Genomes Genet. 2018, 8, 3255–3267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- 1000 Genomes Project. Available online: ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502 (accessed on 2 April 2018).
- Lundmark, P.E.; Liljedahl, U.; Boomsma, D.I.; Mannila, H.; Martin, N.G.; Palotie, A.; Syvänen, A.C. Evaluation of HapMap data in six populations of European descent. Eur. J. Hum. Genet. 2008, 16, 1142–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelis, M.; Esko, T.; Mägi, R.; Zimprich, F.; Zimprich, A.; Toncheva, D.; Karachanak, S.; Piskácková, T.; Balascák, I.; Peltonen, L.; et al. Genetic structure of Europeans: A view from the North-East. PLoS ONE 2009, 4, e5472. [Google Scholar] [CrossRef] [Green Version]
- Khrunin, A.V.; Khokhrin, D.V.; Filippova, I.N.; Esko, T.; Nelis, M.; Bebyakova, N.A.; Bolotova, N.L.; Klovins, J.; Nikitina-Zake, L.; Rehnström, K.; et al. A genome-wide analysis of populations from European Russia reveals a new pole of genetic diversity in northern Europe. PLoS ONE 2013, 8, e58552. [Google Scholar] [CrossRef] [PubMed]
- Khrunin, A.V.; Aliev, A.M.; Limborska, S.A. DNA markers from genome-wide association studies of cardiovascular diseases. Microbiol. Virol. 2018, 33, 245–247. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Ao, S.I.; Yip, K.; Ng, M.; Cheung, D.; Fong, P.Y.; Melhado, I. CLUSTAG: Hierarchical clustering and graph methods for selecting tag SNPs. Bioinformatics 2005, 21, 1735–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bakker, P.I.W.; Yelensky, R.; Pe’Er, I.; Gabriel, S.B.; Daly, M.J.; Altshuler, D. Efficiency and power in genetic association studies. Nat. Genet. 2005, 37, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2004, 21, 263–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.A.; Brossard, M.; Roshandel, D.; Paterson, A.D.; Bull, S.B.; Yoo, Y.J. gpart: Human genome partitioning and visualization of high-density SNP data by identifying haplotype blocks. Bioinformatics 2019, 35, 4419–4421. [Google Scholar] [CrossRef] [PubMed]
- Genotype-Tissue Expression (GTEx) Project. Available online: gtexportal.org. (accessed on 8 July 2020).
- Altenhoff, A.M.; Boeckmann, B.; Capella-Gutierrez, S.; Dalquen, D.A.; DeLuca, T.; Forslund, K. Standardized benchmarking in the quest for orthologs. Nat. Methods 2016, 13, 425–430. [Google Scholar] [CrossRef]
- Nodzak, C. Introductory methods for EQTL analyses. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2020; pp. 3–14. [Google Scholar] [CrossRef]
- Zhao, S.; Jiang, H.; Liang, Z.H.; Ju, H. Integrating Multi-Omics Data to Identify Novel Disease Genes and Single-Neucleotide Polymorphisms. Front. Genet. 2020, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Ma, Y.; Chen, S.; Che, Q.; Chen, D. The Integrated Landscape of Biological Candidate Causal Genes in Coronary Artery. Disease. Front. Genet. 2020, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, M.D.; Chen-Plotkin, A.S. The Post-GWAS Era: From Association to Function. Am. J. Hum. Genet. 2018, 102, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Rat | Human | Metrics | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Chrom | Start (bp) | End (bp) | Gene | Chrom | Start (bp) | End (bp) | 1 | 2 | 3 | 4 | 5 |
| Adora2a | 20 | 16449385 | 16466147 | ADORA2A | 22 | 24813847 | 24838328 | 82 | 82 | 100 | 87.44 | 1 |
| Bcl3 | 1 | 81996116 | 82010351 | BCL3 | 19 | 45250962 | 45263301 | 82 | 83 | 100 | 59.56 | 1 |
| Ccl22 | 19 | 10668403 | 10675173 | CCL22 | 16 | 57392684 | 57400102 | 65 | 65 | 100 | 53.85 | 1 |
| Ccr1 | 8 | 132147929 | 132153481 | CCR1 | 3 | 46243200 | 46249887 | 80 | 80 | 75 | 100 | 1 |
| Cd14 | 18 | 29265353 | 29266946 | CD14 | 5 | 140011313 | 140013286 | 62 | 63 | 75 | 100 | 1 |
| Cd44 | 3 | 99339455 | 99426032 | CD44 | 11 | 35160417 | 35253949 | 71 | 68 | 100 | 91.02 | 1 |
| Csf2rb | 7 | 119544873 | 119558539 | CSF2RB | 22 | 37309670 | 37336491 | 56 | 56 | 100 | 100 | 1 |
| Emp1 | 4 | 233415324 | 233449254 | EMP1 | 12 | 13349650 | 13369708 | 76 | 74 | 75 | 100 | 1 |
| Fosl1 | 1 | 227755887 | 227764393 | FOSL1 | 11 | 65659520 | 65668044 | 92 | 91 | 100 | 100 | 1 |
| Glycam1 | 7 | 142951738 | 142953998 | * | ||||||||
| Gpr6 | 20 | 47518790 | 47521561 | GPR6 | 6 | 110299514 | 110301921 | 94 | 94 | 50 | 99.76 | 1 |
| Gpr88 | 2 | 237334865 | 237339419 | GPR88 | 1 | 101003693 | 101007574 | 95 | 95 | 50 | 100 | 1 |
| Hmox1 | 19 | 25622556 | 25629372 | HMOX1 | 22 | 35776354 | 35790207 | 80 | 80 | 50 | 100 | 1 |
| Il6 | 4 | 3095536 | 3100112 | IL6 | 7 | 22765503 | 22771621 | 40 | 40 | 0 | 100 | 0 |
| Lcn2 | 3 | 16763059 | 16766466 | LCN2 | 9 | 130911350 | 130915734 | 64 | 64 | 100 | 100 | 1 |
| Lgals3 | 15 | 28094062 | 28106276 | LGALS3 | 14 | 55590828 | 55612126 | 82 | 78 | 100 | 96.41 | 1 |
| Mcm5 | 19 | 25637492 | 25681915 | MCM5 | 22 | 35796056 | 35821423 | 47 | 97 | 50 | 99.53 | 0 |
| Olr1 | 4 | 211883405 | 211905489 | OLR1 | 12 | 10310902 | 10324737 | 66 | 49 | 75 | 100 | 0 |
| Osmr | 2 | 75851664 | 75892056 | OSMR | 5 | 38845960 | 38945698 | 56 | 57 | 100 | 99.6 | 1 |
| Ptx3 | 2 | 177457263 | 177463073 | PTX3 | 3 | 157154578 | 157161417 | 81 | 81 | 100 | 100 | 1 |
| Rgs9 | 10 | 97225541 | 97298645 | RGS9 | 17 | 63133549 | 63223821 | 91 | 90 | 75 | 67.67 | 1 |
| Sdc1 | 6 | 43667444 | 43689898 | SDC1 | 2 | 20400558 | 20425194 | 77 | 76 | 100 | 100 | 1 |
| Serpine1 | 12 | 24653385 | 24663763 | SERPINE1 | 7 | 100770370 | 100782547 | 81 | 81 | 100 | 100 | 1 |
| Spp1 | 14 | 6653093 | 6658953 | SPP1 | 4 | 88896819 | 88904562 | 63 | 62 | 100 | 66.28 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khvorykh, G.; Khrunin, A.; Filippenkov, I.; Stavchansky, V.; Dergunova, L.; Limborska, S. A Workflow for Selection of Single Nucleotide Polymorphic Markers for Studying of Genetics of Ischemic Stroke Outcomes. Genes 2021, 12, 328. https://doi.org/10.3390/genes12030328
Khvorykh G, Khrunin A, Filippenkov I, Stavchansky V, Dergunova L, Limborska S. A Workflow for Selection of Single Nucleotide Polymorphic Markers for Studying of Genetics of Ischemic Stroke Outcomes. Genes. 2021; 12(3):328. https://doi.org/10.3390/genes12030328
Chicago/Turabian StyleKhvorykh, Gennady, Andrey Khrunin, Ivan Filippenkov, Vasily Stavchansky, Lyudmila Dergunova, and Svetlana Limborska. 2021. "A Workflow for Selection of Single Nucleotide Polymorphic Markers for Studying of Genetics of Ischemic Stroke Outcomes" Genes 12, no. 3: 328. https://doi.org/10.3390/genes12030328
APA StyleKhvorykh, G., Khrunin, A., Filippenkov, I., Stavchansky, V., Dergunova, L., & Limborska, S. (2021). A Workflow for Selection of Single Nucleotide Polymorphic Markers for Studying of Genetics of Ischemic Stroke Outcomes. Genes, 12(3), 328. https://doi.org/10.3390/genes12030328