
The Genetic and Clinical Features of FOXL2-Related Blepharophimosis, Ptosis and Epicanthus Inversus Syndrome



{kind=link}
{kind=link}
Abstract
:1. Introduction
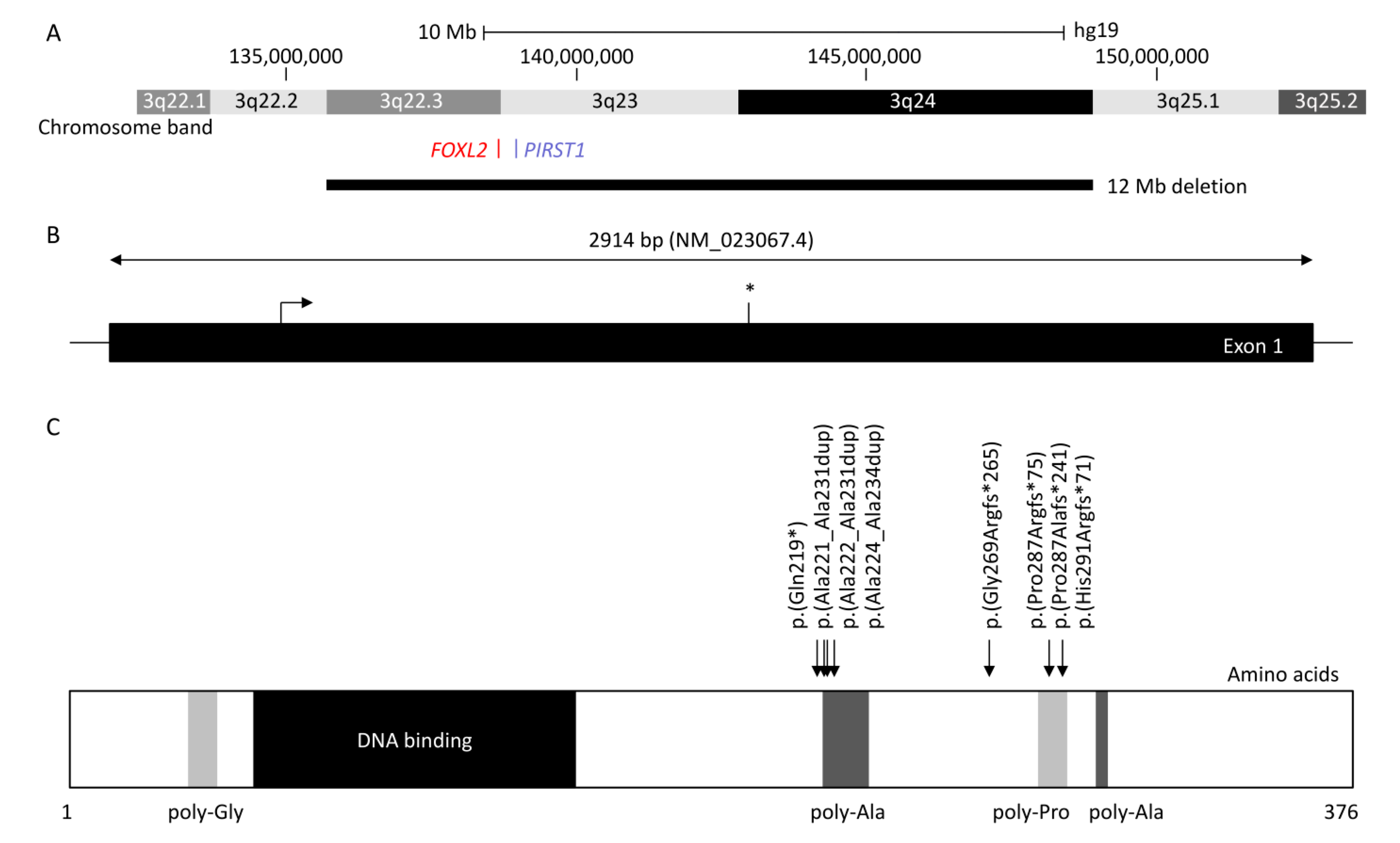
2. Genetics of BPES
2.1. FOXL2 Gene
2.2. Intragenic Variants in FOXL2
2.3. Poly-Alanine Tract Expansion Variants
2.4. Chromosomal Translocations and Involvement of FOXL2 Regulatory Genes
2.5. FOXL2 and Primary Ovarian Failure (POF)
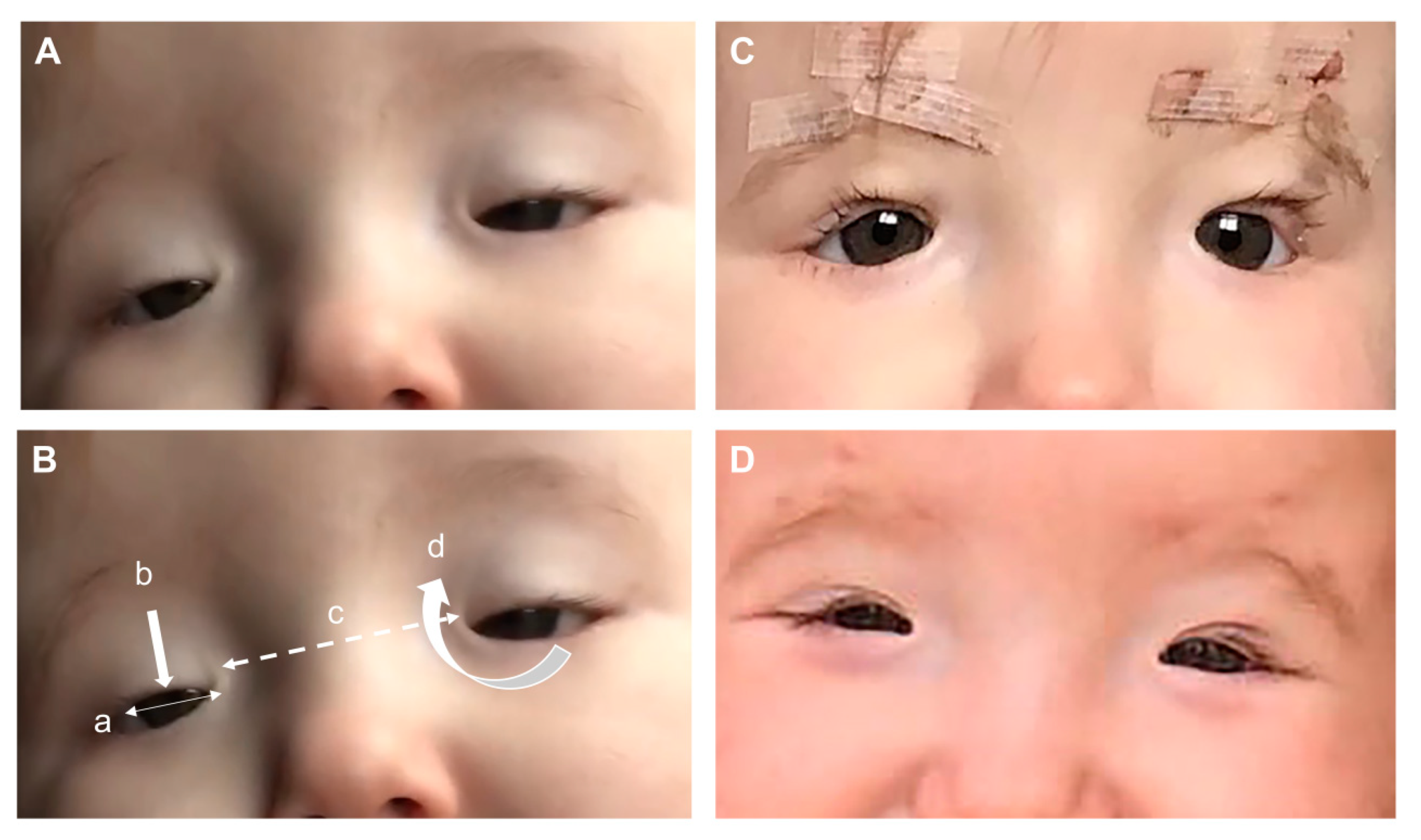
3. Clinical Features
3.1. Differential Diagnoses
3.2. Management
3.3. Genetic Counselling and Testing
3.4. Treatment of Oculofacial Features
3.5. Treatment of Premature Ovarian Failure
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Chawla, B.; Bhadange, Y.; Dada, R.; Kumar, M.; Sharma, S.; Bajaj, M.S.; Pushker, N.; Chandra, M.; Ghose, S. Clinical, Radiologic, and Genetic Features in Blepharophimosis, Ptosis, and Epicanthus Inversus Syndrome in the Indian Population. Investig. Opthalmol. Vis. Sci. 2013, 54, 2985–2991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocquet, J.; Pailhoux, E.; Jaubert, F.; Servel, N.; Xia, X.; Pannetier, M.; De Baere, E.; Messiaen, L.; Cotinot, C.; Fellous, M.; et al. Evolution and Expression of FOXL2. J. Med. Genet. 2002, 39, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Chen, H. Blepharophimosis, Ptosis, and Epicanthus Inversus Syndrome. In Atlas of Genetic Diagnosis and Counseling; Springer: New York, NY, USA, 2012; pp. 233–238. [Google Scholar] [CrossRef]
- Fokstuen, S.; Antonarakis, S.E.; Blouin, J.-L. FOXL2-Mutations in Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome (BPES); Challenges for Genetic Counseling in Female Patients. Am. J. Med. Genet. Part A 2002, 117, 143–146. [Google Scholar] [CrossRef]
- Beysen, D.; Raes, J.; Leroy, B.P.; Lucassen, A.; Yates, J.R.W.; Clayton-Smith, J.; Ilyina, H.; Brooks, S.S.; Christin-Maitre, S.; Fellous, M.; et al. Deletions Involving Long-Range Conserved Nongenic Sequences Upstream and Downstream of FOXL2 as a Novel Disease-Causing Mechanism in Blepharophimosis Syndrome. Am. J. Hum. Genet. 2005, 77, 205–218. [Google Scholar] [CrossRef] [Green Version]
- De Baere, E.; Beysen, D.; Oley, C.; Lorenz, B.; Cocquet, J.; De Sutter, P.; Devriendt, K.; Dixon, M.; Fellous, M.; Fryns, J.-P.; et al. FOXL2 and BPES: Mutational Hotspots, Phenotypic Variability, and Revision of the Genotype-Phenotype Correlation. Am. J. Hum. Genet. 2003, 72, 478–487. [Google Scholar] [CrossRef] [Green Version]
- Batista, F.; Vaiman, D.; Dausset, J.; Fellous, M.; Veitia, R.A. Potential Targets of FOXL2, a Transcription Factor Involved in Craniofacial and Follicular Development, Identified by Transcriptomics. Proc. Natl. Acad. Sci. USA 2007, 104, 3330–3335. [Google Scholar] [CrossRef] [Green Version]
- Leung, D.T.; Fuller, P.J.; Chu, S. Impact of FOXL2 Mutations on Signaling in Ovarian Granulosa Cell Tumors. Int. J. Biochem. Cell Biol. 2016, 72, 51–54. [Google Scholar] [CrossRef]
- Cocquet, J.; De Baere, E.; Gareil, M.; Pannetier, M.; Xia, X.; Fellous, M.; Veitia, R. Structure, Evolution and Expression of the FOXL2 Transcription Unit. Cytogenet. Genome Res. 2003, 101, 206–211. [Google Scholar] [CrossRef] [Green Version]
- D’Haene, B.; Nevado, J.; Pugeat, M.; Pierquin, G.; Lowry, R.; Reardon, W.; Delicado, A.; García-Miñaur, S.; Palomares, M.; Courtens, W.; et al. FOXL2 Copy Number Changes in the Molecular Pathogenesis of BPES: Unique Cohort of 17 Deletions. Hum. Mutat. 2010, 31, E1332–E1347. [Google Scholar] [CrossRef] [PubMed]
- Beysen, D.; De Paepe, A.; De Baere, E. FOXL2 Mutations and Genomic Rearrangements in BPES. Hum. Mutat. 2008, 30, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Alao, M.; Laleye, A.; Lalya, F.; Hans, C.; Abramovicz, M.; Morice-Picard, F.; Arveiler, B.; Lacombe, D.; Rooryck, C. Blepharophimosis, Ptosis, Epicanthus Inversus Syndrome with Translocation and Deletion at Chromosome 3q23 in a Black African female. Eur. J. Med. Genet. 2012, 55, 630–634. [Google Scholar] [CrossRef]
- Bertini, V.; Valetto, A.; Baldinotti, F.; Azzarà, A.; Cambi, F.; Toschi, B.; Giacomina, A.; Gatti, G.L.; Gana, S.; Caligo, M.A.; et al. Blepharophimosis, Ptosis, Epicanthus Inversus Syndrome: New Report with a 197-kb Deletion Upstream of FOXL2 and Review of the Literature. Mol. Syndr. 2019, 10, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Beysen, D.; De Jaegere, S.; Amor, D.; Bouchard, P.; Christin-Maitre, S.; Fellous, M.; Touraine, P.; Grix, A.W.; Hennekam, R.; Meire, F.; et al. Identification of 34 Novel and 56 Known FOXL2 Mutations in Patients with Blepharophimosis Syndrome. Hum. Mutat. 2008, 29, E205–E219. [Google Scholar] [CrossRef] [PubMed]
- Bouman, A.; Van Haelst, M.; Van Spaendonk, R. Blepharophimosis–Ptosis–Epicanthus Inversus Syndrome Caused by a 54-KB Microdeletion in a FOXL2 Cis-Regulatory Element. Clin. Dysmorphol. 2018, 27, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Chacon-Camacho, O.F.; Salgado-Medina, A.; Alcaraz-Lares, N.; López-Moreno, D.; Barragan-Arevalo, T.; Nava-Castañeda, A.; Rodríguez-Uribe, G.; Lieberman, E.; Rodríguez-Cabrera, L.; Angel, A.G.-D.; et al. Clinical Characterization and Identification of Five Novel FOXL2 Pathogenic Variants in a Cohort of 12 Mexican Subjects with the Syndrome of Blepharophimosis-Ptosis-Epicanthus Inversus. Gene 2019, 706, 62–68. [Google Scholar] [CrossRef]
- Chai, P.; Li, F.; Fan, J.; Jia, R.; Zhang, H.; Fan, X. Functional Analysis of a Novel FOXL2 Indel Mutation in Chinese Families with Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome Type, I. Int. J. Biol. Sci. 2017, 13, 1019–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouchene, I.; Derouiche, K.; Chaabouni, A.; Cherif, L.; Amouri, A.; Largueche, L.; Abdelhak, S.; El Matri, L. Identification of a Novel Mutation inFOXL2Gene That Leads to Blepharophimosis Ptosis Epicanthus Inversus and Telecanthus Syndrome in a Tunisian Consanguineous Family. Genet. Test. Mol. Biomarkers 2010, 14, 145–148. [Google Scholar] [CrossRef]
- Corrêa, F.J.S.; Tavares, A.B.; Pereira, R.W.; Abrão, M.S. A New FOXL2 Gene Mutation in a Woman with Premature Ovarian Failure and Sporadic Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome. Fertil. Steril. 2010, 93, 1006.e3–1006.e6. [Google Scholar] [CrossRef] [PubMed]
- Dean, S.J.; Holden, K.R.; Dwivedi, A.; Dupont, B.R.; Lyons, M.J. Acquired Microcephaly in Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome Because of an Interstitial 3q22.3q23 Deletion. Pediatr. Neurol. 2014, 50, 636–639. [Google Scholar] [CrossRef]
- D’Haene, B.; Attanasio, C.; Beysen, D.; Dostie, J.; Lemire, E.; Bouchard, P.; Field, M.; Jones, K.; Lorenz, B.; Menten, B.; et al. Disease-Causing 7.4 kb Cis-Regulatory Deletion Disrupting Conserved Non-Coding Sequences and Their Interaction with the FOXL2 Promotor: Implications for Mutation Screening. PLoS Genet. 2009, 5, e1000522. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Zhou, Y.; Huang, X.; Zhang, L.; Yao, Y.; Song, X.; Chen, J.; Hu, J.; Ge, S.; Song, H.; et al. The Combination of Polyalanine Expansion Mutation and a Novel Missense Substitution in Transcription Factor FOXL2 Leads to Different Ovarian Phenotypes in Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome (BPES) Patients. Hum. Reprod. 2012, 27, 3347–3357. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.-Y.; Han, B.; Qiao, J.; Liu, B.-L.; Ji, Y.-R.; Ge, S.-F.; Song, H.-D.; Fan, X.-Q. Functional Study on a Novel Missense Mutation of the Transcription Factor FOXL2 Causes Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome (BPES). Mutagenesis 2010, 26, 283–289. [Google Scholar] [CrossRef]
- Fan, J.-Y.; Wang, Y.-F.; Han, B.; Ji, Y.-R.; Song, H.-D.; Fan, X.-Q. FOXL2 Mutations in Chinese Families with Blepharophimosis Syndrome (BPES). Transl. Res. 2011, 157, 48–52. [Google Scholar] [CrossRef]
- González-González, C.; Garcia-Hoyos, M.; Calzón, R.H.; Díaz, C.A.; Fanego, C.G.; Sánchez, I.L.; Sánchez-Escribano, F. Microdeletion Found by Array-CGH in Girl with Blepharophimosis Syndrome and Apparently Balanced Translocation t(3;15) (q23;q25). Ophthalmic Genet. 2011, 33, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Gulati, R.; Verdin, H.; Halanaik, D.; Bhat, B.V.; De Baere, E. Co-Occurrence of Congenital Hydronephrosis and FOXL2-Associated Blepharophimosis, Ptosis, Epicanthus Inversus Syndrome (BPES). Eur. J. Med. Genet. 2014, 57, 576–578. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, A.; Verdin, H.; Haghighi-Kakhki, H.; Piri, N.; Gohari, N.S.; De Baere, E. Missense Mutation outside the Forkhead Domain of FOXL2 Causes a Severe Form of BPES Type II. Mol. Vis. 2012, 18, 211–218. [Google Scholar] [PubMed]
- Hu, J.; Ke, H.; Luo, W.; Yang, Y.; Liu, H.; Li, G.; Qin, Y.; Ma, J.; Zhao, S. A novel FOXL2 Mutation in Two Infertile Patients with Blepharophimosis–Ptosis–Epicanthus Inversus Syndrome. J. Assist. Reprod. Genet. 2020, 37, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Guo, J.; Wang, B.; Wang, J.; Zhou, Z.; Zhou, G.; Ding, X.; Ma, X.; Qi, Y. Genetic Analysis of the FOXL2 Gene Using Quantitative Real-Time PCR in Chinese Patients with Blepharophimosis-Ptosis-Epicanthus In-versus Syndrome. Mol. Vis. 2011, 17, 436–442. [Google Scholar]
- Jiang, H.; Huang, X.; Su, Z.; Rao, L.; Wu, S.; Zhang, T.; Li, K.; Quan, Q.; Zhang, K. Genetic Analysis of the Fork-head Transcriptional Factor 2 Gene in Three Chinese Families with Blepharophimosis Syndrome. Mol. Vis. 2013, 19, 418–423. [Google Scholar]
- Kaur, I.; Hussain, A.; Naik, M.N.; Murthy, R.; Honavar, S.G. Mutation spectrum of Fork-Head Transcriptional Factor Gene (FOXL2) in Indian Blepharophimosis Ptosis Epicanthus Inversus Syndrome (BPES) patients. Br. J. Ophthalmol. 2011, 95, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Krepelova, A.; Simandlova, M.; Vlckova, M.; Kuthan, P.; Vincent, A.L.; Liskova, P. Analysis of FOXL2 Detects Three Novel Mutations and an Atypical Phenotype of Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome. Clin. Exp. Ophthalmol. 2016, 44, 757–762. [Google Scholar] [CrossRef]
- Kumar, A.; Babu, M.; Raghunath, A.; Venkatesh, C.P. Genetic Analysis of a Five Generation Indian Family with BPES: A Novel Missense Mutation (p.Y215C). Mol. Vis. 2004, 10, 445–449. [Google Scholar]
- Leon-Mateos, A.; Ginarte, M.; Ruiz-Ponte, C.; Carracedo, A.; Toribio, J. Blepharophimosis Ptosis Epicanthus Inversus Syndrome (BPES). Int. J. Dermatol. 2007, 46, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Chai, P.; Fan, J.; Wang, X.; Lu, W.; Li, J.; Ge, S.; Jia, R.; Zhang, H.; Fan, X. A Novel FOXL2 Mutation Implying Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome Type, I. Cell. Physiol. Biochem. 2018, 45, 203–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Gu, Y. Genetic and Functional Analyses of Two Missense Mutations in the Transcription Factor FOXL2 in Two Chinese Families with Blepharophimosis–Ptosis–Epicanthus Inversus Syndrome. Genet. Test. Mol. Biomarkers 2018, 22, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.C.; Park, W.Y.; Seo, E.-J.; Kim, K.J.; Hwang, Y.S.; Chae, J.H. De Novo Interstitial Deletion of 3q22.3-q25.2 Encompassing FOXL2, ATR, ZIC1, and ZIC4 in a Patient with Blepharophimosis/Ptosis/Epicanthus Inversus Syndrome, Dandy-Walker Malformation, and Global Developmental Delay. J. Child Neurol. 2011, 26, 615–618. [Google Scholar] [CrossRef]
- Lin, W.-D.; Chou, I.-C.; Lee, N.-C.; Wang, C.-H.; Hwu, W.-L.; Lin, S.-P.; Chao, M.-C.; Tsai, Y.; Tsai, F.-J. FOXL2 Mutations in Taiwanese Patients with Blepharophimosis, Ptosis, Epicanthus Inversus Syndrome. Clin. Chem. Lab. Med. 2010, 48, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Aguayo, A.; Poggi, H.; Cattani, A.; Molina, M.; Romeo, E.; Lagos, M. A Novel Insertion in the FOXL2 Gene in a Chilean Patient with Blepharophimosis Ptosis Epicanthus Inversus Syndrome Type I. J. Pediatr. Endocrinol. Metab. 2014, 27, 181–184. [Google Scholar] [CrossRef]
- Méduri, G.; Bachelot, A.; Duflos, C.; Bständig, B.; Poirot, C.; Genestie, C.; Veitia, R.; De Baere, E.; Touraine, P. FOXL2 Mutations Lead to Different Ovarian Phenotypes in BPES Patients: Case Report. Hum. Reprod. 2010, 25, 235–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nallathambi, J.; Moumné, L.; De Baere, E.; Beysen, D.; Usha, K.; Sundaresan, P.; Veitia, R.A. A Novel Polyalanine Expansion in FOXL2: The First Evidence for a Recessive Form of the Blepharophimosis Syndrome (BPES) Associated with Ovarian Dysfunction. Qual. Life Res. 2006, 121, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.K.; Stout, A.U.; Aaby, A.A.; Ng, J.D. Blepharophimosis Syndrome with Absent Tear Production. Ophthalmic Plast. Reconstr. Surg. 2015, 31, e62. [Google Scholar] [CrossRef] [PubMed]
- Ni, F.; Wen, Q.; Wang, B.; Zhou, S.; Wang, J.; Mu, Y.; Ma, X.; Cao, Y. Mutation Analysis of FOXL2 Gene in Chinese Patients with Premature Ovarian Failure. Gynecol. Endocrinol. 2009, 26, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Niu, B.-B.; Tang, N.; Xu, Q.; Chai, P.-W. Genomic Disruption of FOXL2 in Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome Type 2. Chin. Med. J. 2018, 131, 2380–2383. [Google Scholar] [CrossRef] [PubMed]
- Nuovo, S.; Passeri, M.; Di Benedetto, E.; Calanchini, M.; Meldolesi, I.; Di Giacomo, M.C.; Petruzzi, D.; Piemontese, M.R.; Zelante, L.; Sangiuolo, F.; et al. Characterization of Endocrine Features and Genotype–Phenotypes Correlations in Blepharophimosis–Ptosis–Epicanthus Inversus Syndrome Type 1. J. Endocrinol. Investig. 2015, 39, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Raile, K.; Stobbe, H.; Tröbs, R.B.; Kiess, W.; Pfäffle, R. A New Heterozygous Mutation of the FOXL2 Gene Is Associated with a Large Ovarian Cyst and Ovarian Dysfunction in an Adolescent Girl with Blepharophimosis/Ptosis/Epicanthus Inversus Syndrome. Eur. J. Endocrinol. 2005, 153, 353–358. [Google Scholar] [CrossRef]
- Ramineni, A.; Coman, D. De Novo 3q22.3q24 Microdeletion in a Patient with Blepharophimosis–Ptosis–Epicanthus Inversus Syndrome, Dandy-Walker Malformation, and Wisconsin Syndrome. Child Neurol. Open 2016, 3, 2329048–16666362. [Google Scholar] [CrossRef] [Green Version]
- Schlade-Bartusiak, K.; Brown, L.; Lomax, B.; Bruyère, H.; Gillan, T.; Hamilton, S.; McGillivray, B.; Eydoux, P. BPES With Atypical Premature Ovarian Insufficiency, and Evidence of Mitotic Recombination, in a Woman with Trisomy X and a Translocation t(3;11) (q22.3;q14.1). Am. J. Med. Genet. Part A 2012, 158A, 2322–2327. [Google Scholar] [CrossRef]
- Settas, N.; Anapliotou, M.; Kanavakis, E.; Fryssira, H.; Sofocleous, C.; Dacou-Voutetakis, C.; Chrousos, G.P.; Voutetakis, A. A Novel FOXL2 Gene Mutation and BMP15 Variants in a Woman with Primary Ovarian Insufficiency and Blepharophimosis–Ptosis–Epicanthus Inversus Syndrome. Menopause 2015, 22, 1264–1268. [Google Scholar] [CrossRef]
- Tan, H.; Yang, P.; Li, H.; Pan, Q.; Liang, D.; Wu, L. A Novel FOXL2 Mutation in a Chinese Family with Blepharophimosis, Ptosis, Epicanthus Inversus Syndrome. Hum. Genome Var. 2015, 2, 15008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, S.; Wang, X.; Lin, L.; Sun, Y.; Wang, Y.; Yu, H. Mutation Analysis of the FOXL2 Gene in Chinese Patients with Blepharophimosis–Ptosis–Epicanthus Inversus Syndrome. Mutagenesis 2006, 21, 35–39. [Google Scholar] [CrossRef] [Green Version]
- Tzschach, A.; Kelbova, C.; Weidensee, S.; Peters, H.; Ropers, H.-H.; Ullmann, R.; Erdogan, F.; Jurkatis, J.; Menzel, C.; Kalscheuer, V.; et al. Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome in a Girl with Chromosome Translocation t(2;3) (q33;q23). Ophthalmic Genet. 2008, 29, 37–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdin, H.; D’Haene, B.; Beysen, D.; Novikova, Y.; Menten, B.; Sante, T.; Lapunzina, P.; Nevado, J.; Carvalho, C.M.B.; Lupski, J.R.; et al. Microhomology-Mediated Mechanisms Underlie Non-Recurrent Disease-Causing Microdeletions of the FOXL2 Gene or Its Regulatory Domain. PLoS Genet. 2013, 9, e1003358. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, J.; Zhang, Q. FOXL2 Mutations in Chinese Patients with Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome. Mol. Vis. 2007, 13, 108–113. [Google Scholar]
- Xu, Y.; Lei, H.; Dong, H.; Zhang, L.; Qin, Q.; Gao, J.; Zou, Y.; Yan, X. FOXL2 Gene Mutations and Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome (BPES): A Novel Mutation Detected in a Chinese Family and a Statistic Model for Summarizing Previous Reported Records. Mutagenesis 2009, 24, 447–453. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Zheng, J.; Zhou, Q.; Hejtmancik, J.F.; Wang, Y.; Li, S. Novel FOXL2 Mutations in Two Chinese Families with Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome. BMC Med. Genet. 2015, 16, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Li, T.; Xing, Y. Identification of a Novel FOXL2 Mutation in a Single Family with Both Types of Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome. Mol. Med. Rep. 2017, 16, 5529–5532. [Google Scholar] [CrossRef]
- Yang, X.-W.; He, W.-B.; Gong, F.; Li, W.; Li, X.-R.; Zhong, C.-G.; Lu, G.-X.; Lin, G.; Du, J.; Tan, Y.-Q. Novel FOXL2 Mutations Cause Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome with Premature Ovarian Insufficiency. Mol. Genet. Genom. Med. 2018, 6, 261–267. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yang, C.; Zhu, Y.; Chen, H.; Zhao, R.; He, X.; Tao, L.; Wang, P.; Zhou, L.; Zhao, L.; et al. Intragenic and Extragenic Disruptions of FOXL2 Mapped by Whole Genome Low-Coverage Sequencing in Two BPES Families with Chromosome Reciprocal Translocation. Genomes 2014, 104, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Zahanova, S.; Meaney, B.; Łabieniec, B.; Verdin, H.; De Baere, E.; Nowaczyk, M.J. Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome Plus. Clin. Dysmorphol. 2012, 21, 48–52. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, L.; Han, R.; Guan, L.; Fan, B.; Liu, M.; Ying, M.; Peng, H.; Li, N. Identification of the Forkhead Transcriptional Factor 2 (FOXL2) Gene Mutations in Four Chinese Families with Blepharophimosis Syndrome. Mol. Vis. 2013, 19, 2298–2305. [Google Scholar]
- Zhou, L.; Wang, J.; Wang, T. Functional Study on New FOXL2 Mutations Found in Chinese Patients with Blepharophimosis, Ptosis, Epicanthus Inversus Syndrome. BMC Med. Genet. 2018, 19, 121. [Google Scholar] [CrossRef] [Green Version]
- Bunyan, D.J.; Thomas, N.S. Screening of a Large Cohort of Blepharophimosis, Ptosis, and Epicanthus Inversus Syndrome Patients Reveals a Very Strong PA-Ternal Inheritance Bias and a Wide Spectrum of Novel FOXL2 Mutations. Eur. J. Med. Genet. 2019, 62, 103668. [Google Scholar] [CrossRef]
- Brocke, K.S.; Neu-Yilik, G.; Gehring, N.H.; Hentze, M.W.; Kulozik, A.E. The Human Intronless Melanocortin 4-Receptor Gene Is NMD Insensitive. Hum. Mol. Genet. 2002, 11, 331–335. [Google Scholar] [CrossRef] [Green Version]
- Moumné, L.; Fellous, M.; Veitia, R.A. Deletions in the Polyalanine-Containing Transcription Factor FOXL2 Lead to Intranuclear Aggregation. Hum. Mol. Genet. 2005, 14, 3557–3564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Baere, E.; Dixon, M.J.; Small, K.W.; Jabs, E.W.; Leroy, B.P.; Devriendt, K.; Gillerot, Y.; Mortier, G.; Meire, F.; Van Maldergem, L.; et al. Spectrum of FOXL2 Gene Mutations in Blepharophimosis-Ptosis-Epicanthus Inversus (BPES) Families Demonstrates a Genotype-Phenotype Correla-Tion. Hum. Mol. Genet. 2001, 10, 1591–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, L.Y.; Brown, S.A. Alanine Tracts: The Expanding Story of Human Illness and Trinucleotide Repeats. Trends Genet. 2004, 20, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Kosaki, K.; Ogata, T.; Kosaki, R.; Sato, S.; Matsuo, N. A Novel Mutation in the FOXL2 Gene in a Patient with Blepharophimosis Syndrome: Differential Role of the Polyalanine Tract in the Development of the Ovary and the Eyelid. Ophthalmic Genet. 2002, 23, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Caburet, S.; Demarez, A.; Moumné, L.; Fellous, M.; De Baere, E.A.; Veitia, R. A Recurrent Polyalanine Expansion in the Transcription Factor FOXL2 Induces Extensive Nuclear and Cytoplasmic Protein Aggregation. J. Med. Genet. 2004, 41, 932–936. [Google Scholar] [CrossRef] [Green Version]
- Moumné, L.; Dipietromaria, A.; Batista, F.; Kocer, A.; Fellous, M.; Pailhoux, E.; Veitia, R.A. Differential Aggregation and Functional Impairment Induced by Polyalanine Expansions in FOXL2, a Transcription Factor in-Volved in Cranio-Facial and Ovarian Development. Hum. Mol. Genet. 2007, 17, 1010–1019. [Google Scholar] [CrossRef] [Green Version]
- Boccone, L.; Meloni, A.; Falchi, A.M.; Usai, V.; Cao, A. Blepharophimosis, Ptosis, Epicanthus Inversus Syndrome, a New Case Associated with de Novo Balanced Autosomal Translocation [46,XY,t(3;7)(q23;q32)]. Am. J. Med. Genet. 1994, 51, 258–259. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, Y.; Wakui, K.; Nishida, T.; Ueoka, Y. Blepharophimosis Sequence Andde Novo Balanced Autosomal Translocation [46, XY,t(3;4)(q23;p15.2)]: Possible Assignment of the Trait to 3q23. Am. J. Med. Genet. 1991, 40, 485–487. [Google Scholar] [CrossRef]
- Kleinjan, D.-J.; Van Heyningen, V. Position Effect in Human Genetic Disease. Hum. Mol. Genet. 1998, 7, 1611–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikic, S.; Vaiman, D. Conserved Patterns of Gene Expression in Mice and Goats in the Vicinity of the Polled Intersex Syndrome (PIS) locus. Chromosom. Res. 2004, 12, 465–474. [Google Scholar] [CrossRef]
- Pailhoux, E.; Vigier, B.; Schibler, L.; Cribiu, E.P.; Cotinot, C.; Vaiman, D. Positional Cloning of the PIS Mutation in Goats and Its Impact on Understanding Mammalian Sex-Differentiation. Genet. Sel. Evol. 2005, 37, S55. [Google Scholar] [CrossRef]
- Uhlenhaut, N.H.; Treier, M. FOXL2 Function in Ovarian Development. Mol. Genet. Metab. 2006, 88, 225–234. [Google Scholar] [CrossRef]
- Sills, E.S.; Alper, M.M.; Walsh, A.P. Ovarian Reserve Screening in Infertility: Practical Applications and Theoretical Directions for Research. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009, 146, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.L.; Chabbert-Buffet, N.; Darai, E. Diminished Ovarian Reserve, Premature Ovarian Failure, Poor Ovarian Responder—A Plea for Universal Definitions. J. Assist. Reprod. Genet. 2015, 32, 1709–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastore, L.M.; Christianson, M.S.; Stelling, J.; Kearns, W.G.; Segars, J.H. Reproductive Ovarian Testing and the Alphabet Soup of Diagnoses: DOR, POI, POF, POR, and FOR. J. Assist. Reprod. Genet. 2018, 35, 17–23. [Google Scholar] [CrossRef]
- Roth, L.; Alvero, R. Pregnancy in a Woman with Premature Ovarian Insufficiency Associated with Blepharo-phimosis, Ptosis, Epicanthus Inversus Syndrome Type I: A Case Report. Request PDF. J. Reprod. Med. 2014, 59, 87–89. [Google Scholar] [PubMed]
- Beaconsfield, M.; Walker, J.W.; Collin, J.R. Visual Development in the Blepharophimosis Syndrome. Br. J. Ophthalmol. 1991, 75, 746–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyers, A.G. The Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome (BPES). Orbit 2011, 30, 199–201. [Google Scholar] [CrossRef] [PubMed]
- Verdin, H.; Baere, E. De Blepharophimosis, Ptosis, and Epicanthus Inversus; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; 8 Jul. 2004 [updated 5 Feb. 2015]; GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 1993–2020. Available online: https://pubmed.ncbi.nlm.nih.gov/20301614/ (accessed on 30 January 2021).
- Drury, S.; Hill, M.; Chitty, L. Cell-Free Fetal DNA Testing for Prenatal Diagnosis. In Advances in Applied Microbiology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 76, pp. 1–35. [Google Scholar] [CrossRef]
- Richardson, R.; Smart, M.; Tracey-White, D.; Webster, A.R.; Moosajee, M. Mechanism and Evidence of Nonsense Suppression Therapy for Genetic Eye Disorders. Exp. Eye Res. 2017, 155, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Fuller-Carter, P.I.; Basiri, H.; Harvey, A.R.; Carvalho, L.S. Focused Update on AAV-Based Gene Therapy Clinical Trials for Inherited Retinal Degeneration. BioDrugs 2020, 34, 763–781. [Google Scholar] [CrossRef]
- Beckingsale, P.S.; Sullivan, T.J.; Wong, V.A.; Oley, C. Blepharophimosis: A Recommendation for Early Surgery in Patients with Severe Ptosis. Clin. Exp. Ophthalmol. 2003, 31, 138–142. [Google Scholar] [CrossRef]
- Dawson, E.; Hardy, T.; Collin, J.; Lee, J. The Incidence of Strabismus and Refractive Error in Patients with Blepharophimosis, Ptosis and Epicanthus Inversus Syndrome (BPES). Strabismus 2003, 11, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, Q.; Li, L.; Liu, W.; Li, C.; Fan, Y.; Cao, W.; Li, N. A Modified Fox Pentagon Technique Performed Using a Polytetrafluoroethylene Sling in Frontalis Suspension to Treat Blepharophimosis Syndrome. Sci. Prog. 2020, 103, 003685041989388. [Google Scholar] [CrossRef] [Green Version]
- Al-Abbadi, Z.; Sagili, S.; Malhotra, R. Outcomes of Posterior-Approach ‘Levatorpexy’ in Congenital Ptosis Repair. Br. J. Ophthalmol. 2014, 98, 1686–1690. [Google Scholar] [CrossRef]
- Antus, Z.; Salam, A.; Horvath, E.; Malhotra, R. Outcomes for Severe Aponeurotic Ptosis Using Posterior Approach White-Line Advancement Ptosis Surgery. Eye 2017, 32, 81–86. [Google Scholar] [CrossRef]
- Taylor, A.; Strike, P.W.; Tyers, A.G. Blepharophimosis Ptosis Epicanthus Inversus Syndrome: Objective Analysis of Surgical Outcome in Patients from a Single Unit. Clin. Exp. Ophthalmol. 2007, 35, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Shao, Y.; Zhao, Z.; Zhang, D. One-Stage Correction of Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome Using a Frontalis Muscle Transfer Technique. J. Plast. Surg. Hand Surg. 2013, 48, 74–79. [Google Scholar] [CrossRef]
- Bhattacharjee, K.; Bhattacharjee, H.; Kuri, G.; Shah, Z.T.; Deori, N. Single Stage Surgery for Blepharophimosis Syndrome. Indian J. Ophthalmol. 2012, 60, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Sebastiá, R.; Neto, G.H.; Fallico, E.; Lessa, S.; Solari, H.P.; Ventura, M.P. A One-Stage Correction of the Blepharophimosis Syndrome Using a Standard Combination of Surgical Techniques. Aesthetic Plast. Surg. 2011, 35, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Parvizi, S.; Ong, J.; Rayyah, Y.A.; Dunaway, D. A Novel Medial Canthal Reconstruction Technique in Children with Blepharophimosis Syndrome. Ophthalmic Plast. Reconstr. Surg. 2019, 35, 506–508. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Jia, R.; Zhu, H.; Zhou, Y.; Sun, Y.; Lin, M.; Fu, Y.; Li, J.; Li, Z.; Lu, L.; et al. A Modified Staged Surgical Intervention for Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome. Ann. Plast. Surg. 2015, 74, 410–417. [Google Scholar] [CrossRef]
- Mustardé, J. Epicanthus and Telecanthus. Br. J. Plast. Surg. 1963, 16, 346–356. [Google Scholar] [CrossRef]
- Goswami, D.; Conway, G.S. Premature Ovarian Failure. Hum. Reprod. Updat. 2005, 11, 391–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Méjécase, C.; Nigam, C.; Moosajee, M.; Bladen, J.C. The Genetic and Clinical Features of FOXL2-Related Blepharophimosis, Ptosis and Epicanthus Inversus Syndrome. Genes 2021, 12, 364. https://doi.org/10.3390/genes12030364
Méjécase C, Nigam C, Moosajee M, Bladen JC. The Genetic and Clinical Features of FOXL2-Related Blepharophimosis, Ptosis and Epicanthus Inversus Syndrome. Genes. 2021; 12(3):364. https://doi.org/10.3390/genes12030364
Chicago/Turabian StyleMéjécase, Cécile, Chandni Nigam, Mariya Moosajee, and John C. Bladen. 2021. "The Genetic and Clinical Features of FOXL2-Related Blepharophimosis, Ptosis and Epicanthus Inversus Syndrome" Genes 12, no. 3: 364. https://doi.org/10.3390/genes12030364
APA StyleMéjécase, C., Nigam, C., Moosajee, M., & Bladen, J. C. (2021). The Genetic and Clinical Features of FOXL2-Related Blepharophimosis, Ptosis and Epicanthus Inversus Syndrome. Genes, 12(3), 364. https://doi.org/10.3390/genes12030364