
Simultaneous Homozygous Mutations in SLC12A3 and CLCNKB in an Inbred Chinese Pedigree

Abstract
:1. Introduction
2. Materials and Methods
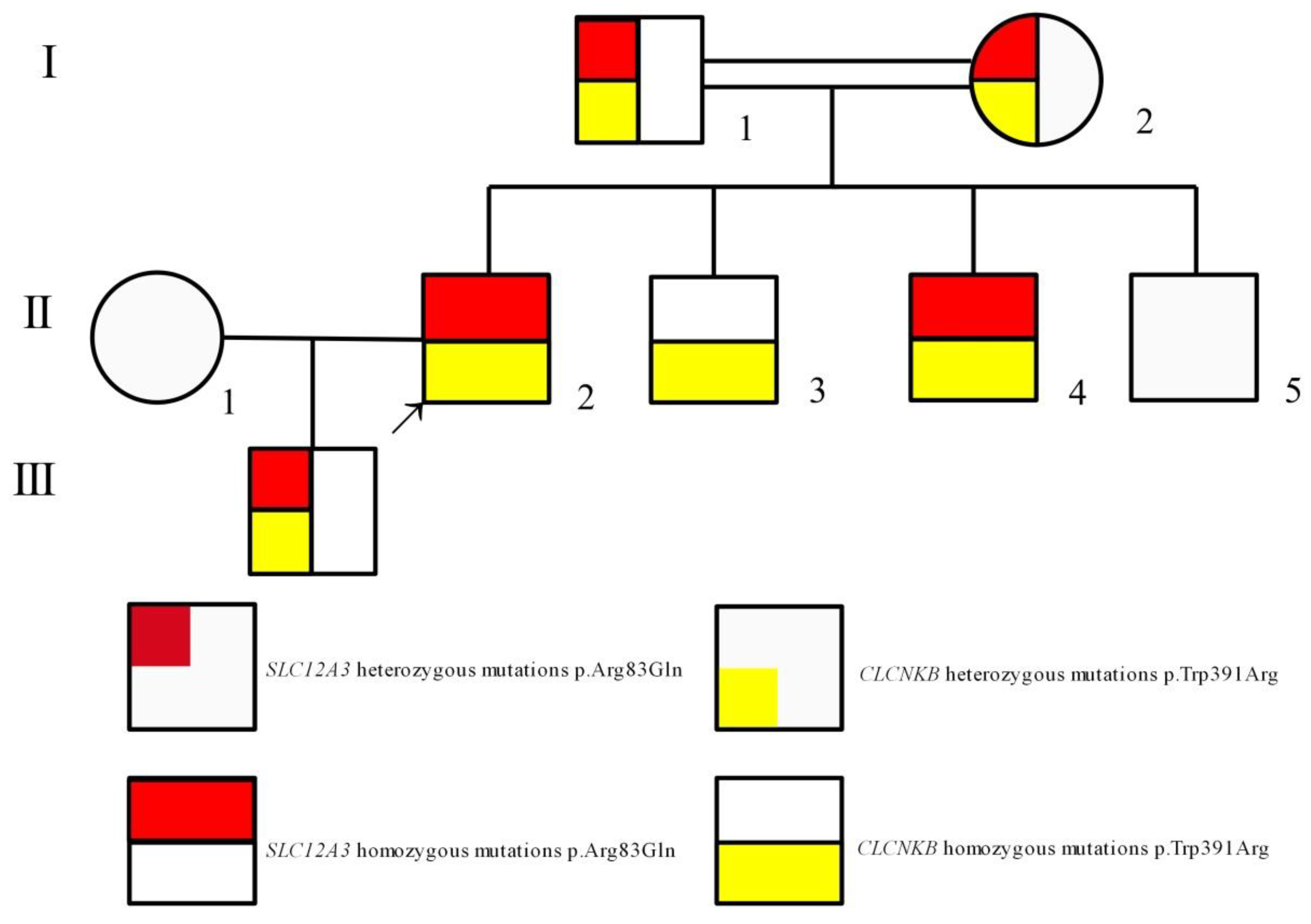
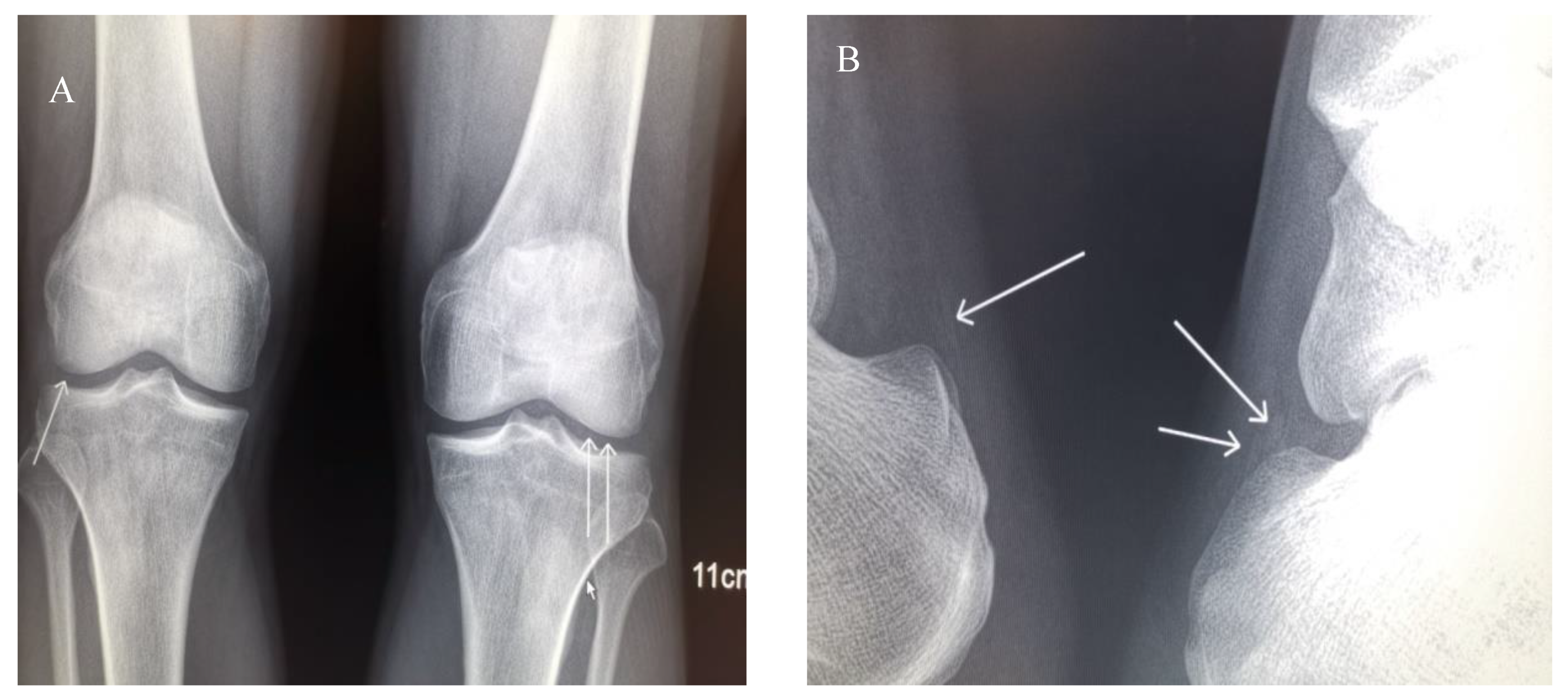
2.1. Case Description
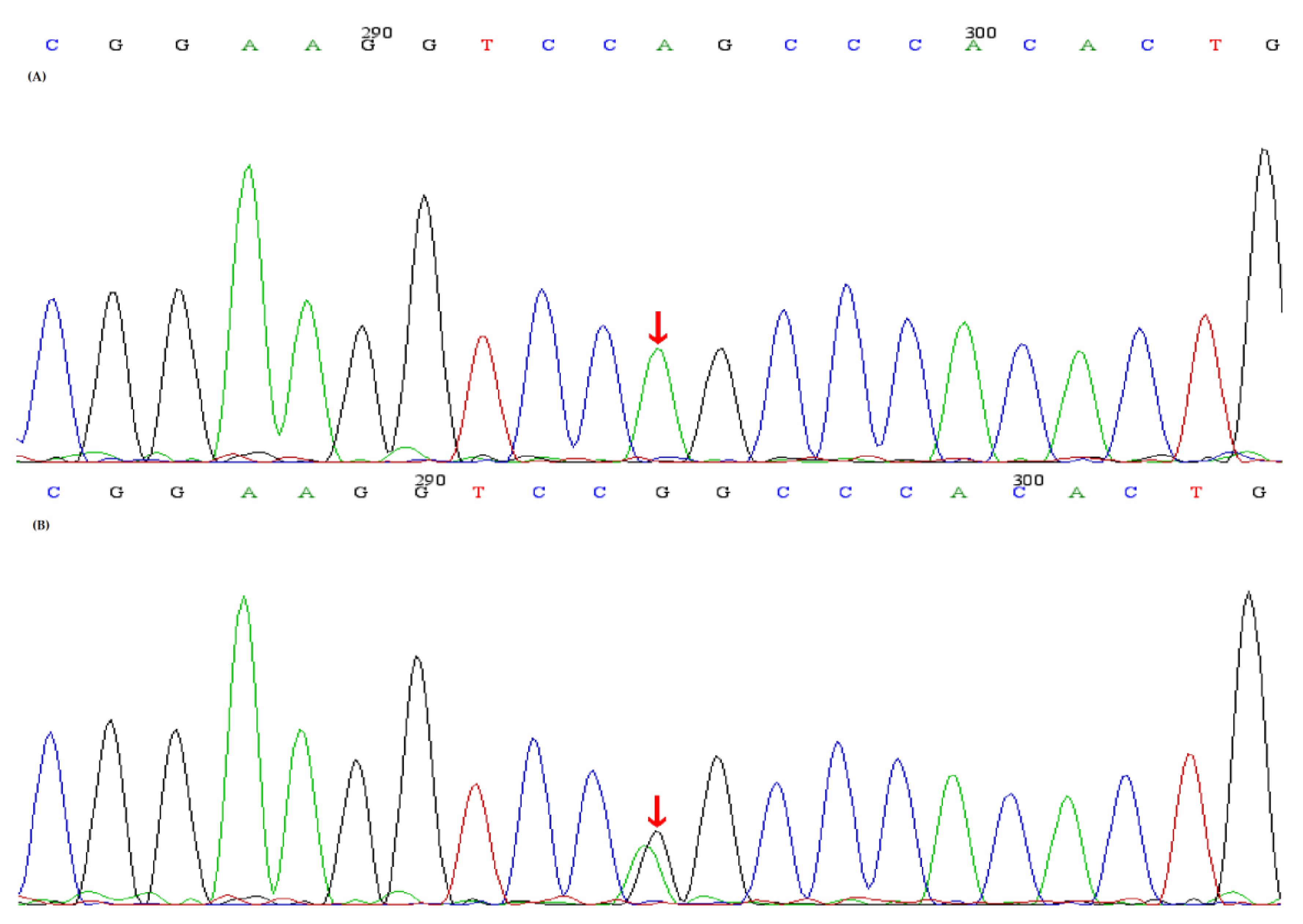
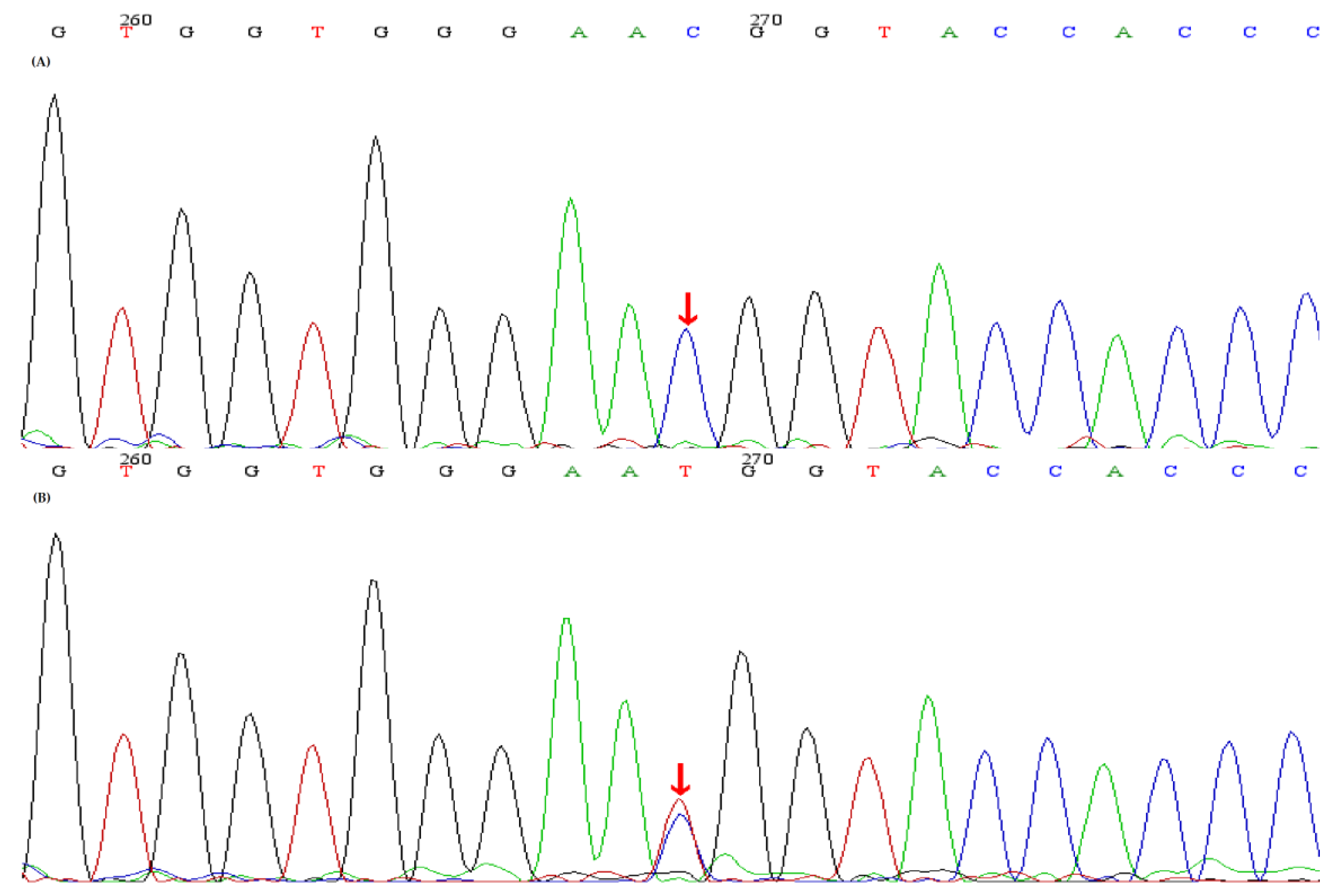
2.2. Genetic Analysis
2.3. Treatment and Follow Up
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nozu, K.; Iijima, K.; Kanda, K.; Nakanishi, K.; Yoshikawa, N.; Satomura, K.; Kaito, H.; Hashimura, Y.; Ninchoji, T.; Komatsu, H.; et al. The Pharmacological Characteristics of Molecular-Based Inherited Salt-Losing Tubulopathies. J. Clin. Endocrinol. Metab. 2010, 95, E511–E518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennings, J.C.; Andrini, O.; Picard, N.; Paulais, M.; Huebner, A.K.; Cayuqueo, I.K.; Bignon, Y.; Keck, M.; Cornière, N.; Böhm, D.; et al. The ClC-K2 Chloride Channel Is Critical for Salt Handling in the Distal Nephron. J. Am. Soc. Nephrol. 2017, 28, 209–217. [Google Scholar] [CrossRef]
- Blanchard, A.; Bockenhauer, D.; Bolignano, D.; Calò, L.A.; Cosyns, E.; Devuyst, O.; Ellison, D.H.; Frankl, F.E.K.; Knoers, N.V.; Konrad, M.; et al. Gitelman syndrome: Consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2017, 91, 24–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seys, E.; Andrini, O.; Keck, M.; Mansour-Hendili, L.; Courand, P.-Y.; Simian, C.; Deschenes, G.; Kwon, T.; Bertholet-Thomas, A.; Bobrie, G.; et al. Clinical and Genetic Spectrum of Bartter Syndrome Type 3. J. Am. Soc. Nephrol. 2017, 28, 2540–2552. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Doherty, M.; Bardini, T.; Barskova, V.; Guerne, P.-A.; Jansen, T.L.; Leeb, B.F.; Perez-Ruiz, F.; Pimentao, J.; Punzi, L.; et al. European League Against Rheumatism recommendations for calcium pyrophosphate deposition. Part I: Terminology and diagnosis. Ann. Rheum. Dis. 2011, 70, 563–570. [Google Scholar] [CrossRef]
- Favero, M.; Calo, L.A.; Schiavon, F.; Punzi, L. Miscellaneous non-inflammatory musculoskeletal conditions. Bartter’s and Gitelman’s diseases. Best Pract. Res. Clin. Rheumatol. 2011, 25, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Meyer, W.J., 3rd; Gill, J.R., Jr.; Bartter, F.C. Gout as a complication of Bartter’s syndrome. A possible role for alkalosis in the decreased clearance of uric acid. Ann. Intern. Med. 1975, 83, 56–59. [Google Scholar] [CrossRef]
- Vargas-Poussou, R.; Dahan, K.; Kahila, D.; Venisse, A.; Riveira-Munoz, E.; Debaix, H.; Grisart, B.; Bridoux, F.; Unwin, R.; Moulin, B.; et al. Spectrum of Mutations in Gitelman Syndrome. J. Am. Soc. Nephrol. 2011, 22, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Uchida, S.; Okamura, H.-O.; Marumo, F.; Sasaki, S. Human CLC-KB gene promoter drives the EGFP expression in the specific distal nephron segments and inner ear. J. Am. Soc. Nephrol. 2002, 13, 1992–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelikovic, I.; Szargel, R.; Hawash, A.; Labay, V.; Hatib, I.; Cohen, N.; Nakhoul, F. A novel mutation in the chloride channel gene, CLCNKB, as a cause of Gitelman and Bartter syndromes. Kidney Int. 2003, 63, 24–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettinelli, A.; Borsa, N.; Syrén, M.-L.; Mattiello, C.; A Coviello, D.; Edefonti, A.; Giani, M.; Travi, M.; Tedeschi, S. Simultaneous Mutations in the CLCNKB and SLC12A3 Genes in Two Siblings with Phenotypic Heterogeneity in Classic Bartter Syndrome. Pediatr. Res. 2005, 58, 1269–1273. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Xu, K.; Yuan, K.; Zhu, J.; Gu, W.; Liang, L.; Wang, C. Digenetic inheritance of SLC12A3 and CLCNKB genes in a Chinese girl with Gitelman syndrome. BMC Pediatr. 2019, 19, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Lee, J.; Heo, N.J.; Cheong, H.I.; Han, J.S. Mutations in SLC12A3 and CLCNKB and Their Correlation with Clinical Phenotype in Patients with Gitelman and Gitelman-like Syndrome. J. Korean Med Sci. 2016, 31, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konrad, M.; Vollmer, M.; Lemmink, H.H.; Heuvel, L.P.V.D.; Jeck, N.; Vargas-Poussou, R.; Lakings, A.; Ruf, R.; Deschênes, G.; Antignac, C.; et al. Mutations in the chloride channel gene CLCNKB as a cause of classic Bartter syndrome. J. Am. Soc. Nephrol. 2000, 11, 1449–1459. [Google Scholar] [PubMed]
- Riveira-Munoz, E.; Chang, Q.; Godefroid, N.; Hoenderop, J.G.; Bindels, R.J.; Dahan, K.; Devuyst, O. Transcriptional and Functional Analyses ofSLC12A3Mutations: New Clues for the Pathogenesis of Gitelman Syndrome. J. Am. Soc. Nephrol. 2007, 18, 1271–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, E.; Cristóbal, P.S.; Rivera, M.; Vázquez, N.; Bobadilla, N.A.; Gamba, G. Affinity-defining domains in the Na-Cl cotransporter: A different location for Cl− and thiazide binding. J. Biol. Chem. 2006, 281, 17266–17275. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject | I-1 | I-2 | II-2 | II-3 | II-4 | II-5 | III-1 | Reference Range |
|---|---|---|---|---|---|---|---|---|
| Age (year) | 69 | 69 | 48 | 46 | 44 | 42 | 25 | |
| Sex | Male | Female | Male | Male | Male | Male | Male | |
| Serum biochemisty | ||||||||
| Cr (μmol/L) | 115 | 77 | 99 | 92 | 129 | 93 | 117 | 62~115 |
| UAa (μmol/L) | 605 | 288 | 998 | 433 | 324 | 458 | 582 | 150~420 |
| Na (mmol/L) | 142 | 140 | 138 | 140 | 133 | 139 | 140 | 137~147 |
| Cl (mmol/L) | 101 | 100 | 93 | 99 | 88 | 102 | 101 | 99~110 |
| K (mmol/L) | 3.4 | 3.9 | 2.6 | 3.0 | 2.4 | 4.1 | 4.2 | 3.5~5.3 |
| Mg (mmol/L) | 0.99 | 0.92 | 0.53 | 0.95 | 0.79 | 0.96 | 0.91 | 0.66~1.07 |
| Ca (mmol/L) | 2.26 | 2.38 | 2.15 | 2.13 | 2.33 | 2.19 | 2.33 | 2.0~2.50 |
| eGFR b (mL/min/1.73 m2) | 56.02 | 67.87 | 77.27 | 85.63 | 57.71 | 86.92 | 74.21 | >90 |
| Urine analysis | ||||||||
| Specific gravity | 1.015 | 1.010 | 1.010 | 1.010 | 1.010 | 1.020 | 1.015 | 1.003~1.030 |
| pH | 5.5 | 5.50 | 7 | 6.5 | 7.5 | 5.5 | 5.5 | 4.5~8.0 |
| Red blood cell (cells/μL) | 111 | 0 | 4 | 1 | 1 | 8 | 3 | 0~15 |
| Protein | 0 | 0 | 0 | 0 | 0 | 0 | 0 | Negative |
| 24-h urine | ||||||||
| Urine volume (mL/24 h) | 1450 | 750 | 3500 | 1600 | 1800 | 1700 | 1200 | |
| K (mmol/24 h) | 32.77 | 23.1 | 47.6 | 47.84 | 73.98 | 17.85 | 24.84 | |
| Ca (mmol/24 h) | 0.7 | 8.192 | 1.26 | |||||
| Spot Ca/Cr ratio (mmol/mmol) | 0.23 | 0.56 | 0.01 | 0.63 | 0.05 | 0.34 | 0.04 | |
| Plasma Renin activity (upright) (ng/mL/h) | 15.95 | 3.37 | 7.18 | 0.93~6.56 | ||||
| Plasma Aldosterone (upright) (pg/mL) | 37.36 | 215.38 | 526.67 | 65.0~296.0 | ||||
| Arterial blood gas | ||||||||
| pH | 7.49 | 7.41 | 7.47 | 7.35~7.45 | ||||
| PaO2 (mmHg) | 74 | 105 | 104 | 80~100 | ||||
| PaCO2 (mmHg) | 37 | 36.8 | 40.8 | 35~45 | ||||
| HCO3- (mmol/L) | 27.9 | 22.8 | 29.6 | 21~28 | ||||
| BE c (mmol/L) | 4.9 | −1.1 | 5.9 | +3~−3 | ||||
| Ultrasound of urinary tract | Left kidney stone | Normal | Normal | Normal | Bilateral kidney stones and right cyst | Bilateral kidney stones | Normal |
| Gene | Exon | Transcript | Nucleotide Mutations | Amino Acid Variants | Variant Type | PolyPhen-2 | Mutation Taster | SIFT | GERP |
|---|---|---|---|---|---|---|---|---|---|
| SLC12A3 | 1 | NM_000085.5 | c.248G>A | p.Arg83Gln | missense | 1 (probably-damaging) | 1 (disease-causing) | 0 (damaging) | 5.42 (conserved) |
| CLCNKB | 12 | NM_000339.3 | c.1171T>C | p.Trp391Arg | missense | 0.999 (probably-damaging) | 1 (disease-causing) | 0 (damaging) | 4.59 (conserved) |
| SIFT: Sorting Intolerant From Tolerant, GERP: Gnomic Evolutionary Rate Profiling | |||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mou, L.; Wu, F. Simultaneous Homozygous Mutations in SLC12A3 and CLCNKB in an Inbred Chinese Pedigree. Genes 2021, 12, 369. https://doi.org/10.3390/genes12030369
Mou L, Wu F. Simultaneous Homozygous Mutations in SLC12A3 and CLCNKB in an Inbred Chinese Pedigree. Genes. 2021; 12(3):369. https://doi.org/10.3390/genes12030369
Chicago/Turabian StyleMou, Lijun, and Fengfen Wu. 2021. "Simultaneous Homozygous Mutations in SLC12A3 and CLCNKB in an Inbred Chinese Pedigree" Genes 12, no. 3: 369. https://doi.org/10.3390/genes12030369
APA StyleMou, L., & Wu, F. (2021). Simultaneous Homozygous Mutations in SLC12A3 and CLCNKB in an Inbred Chinese Pedigree. Genes, 12(3), 369. https://doi.org/10.3390/genes12030369