
The Role of Epigenetics in Congenital Heart Disease



Abstract
:1. Introduction
2. Overview of Congenital Heart Disease
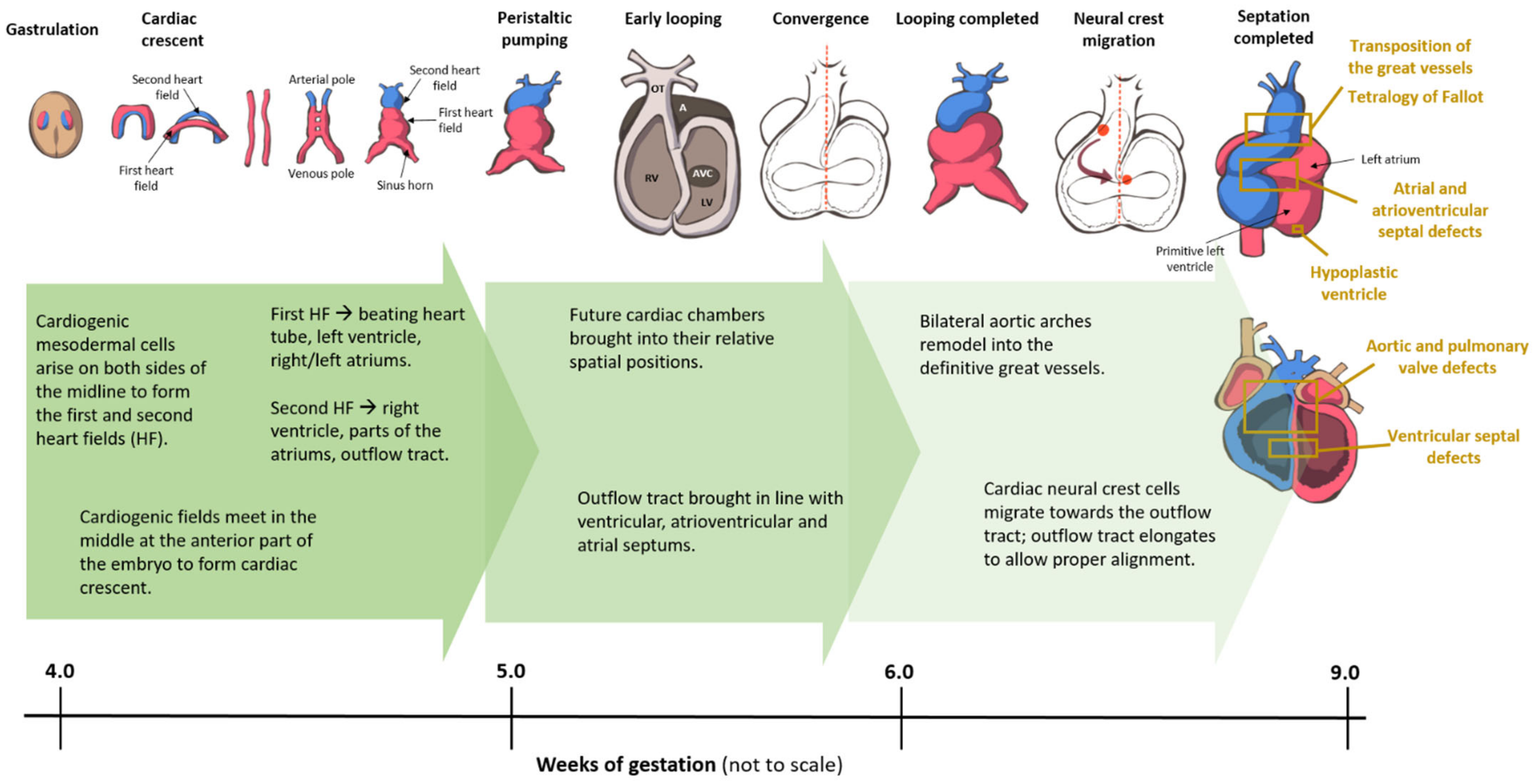
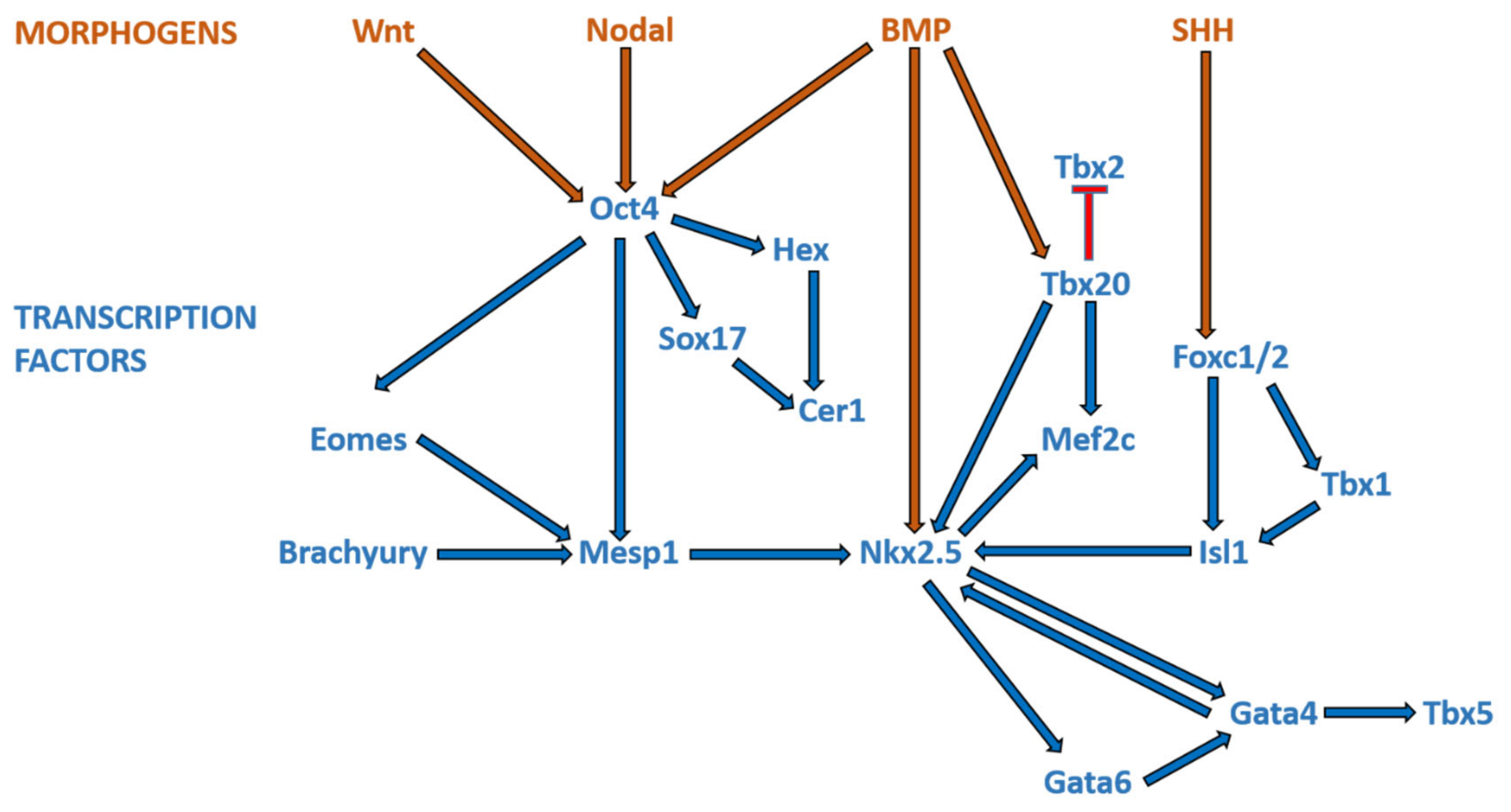
3. Overview of Cardiogenesis and Transcriptional Events during Cardiac Development
3.1. Formation of the Linear Heart Tube
3.2. Cardiac Looping
3.3. Septation
3.3.1. Atrial and Atrioventricular Canal Septation
3.3.2. Ventricular Septation
3.3.3. Outflow Tract Septation
4. Epigenetics and Congenital Heart Disease
4.1. DNA Methylation
DNA Methylation and CHD
4.2. Histone Modifications and Chromatin Modeling
4.2.1. Histone Modifications and CHD
4.2.2. Chromatin-Remodeling Complexes and CHD
4.3. Non-Coding RNA
4.3.1. The Roles of Non-Coding RNA in Cardiac Differentiation
4.3.2. Non-Coding RNA and CHD
MicroRNAs and TOF/Cyanotic CHD
MicroRNAs and Septal Defects
lncRNAs and CHD-
circRNAs and CHD
4.4. Other Epigenetic Mechanisms
4.4.1. Sumoylation
4.4.2. RNA Modifications
5. Current Challenges and Future Directions
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hoffman, J.I.; Kaplan, S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002, 39, 1890–1900. [Google Scholar] [CrossRef] [Green Version]
- Fahed, A.C.; Gelb, B.D.; Seidman, J.G.; Seidman, C.E. Genetics of congenital heart disease: The glass half empty. Circ. Res. 2013, 112, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Agopian, A.J.; Goldmuntz, E.; Hakonarson, H.; Sewda, A.; Taylor, D.; Mitchell, L.E. Pediatric Cardiac Genomics Consortium* Genome-Wide Association Studies and Meta-Analyses for Congenital Heart Defects. Circ. Cardiovasc. Genet. 2017, 10, e001449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerman, M.S.; Smith, A.G.C.; Sable, C.A.; Echko, M.M.; Wilner, L.B.; Olsen, H.E.; Atalay, H.T.; Awasthi, A.; Bhutta, Z.A.; Boucher, J.L.; et al. Global, regional, and national burden of congenital heart disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Child Adolesc. Health 2020, 4, 185–200. [Google Scholar] [CrossRef] [Green Version]
- Marelli, A.J.; Ionescu-Ittu, R.; Mackie, A.S.; Guo, L.; Dendukuri, N.; Kaouache, M. Lifetime Prevalence of Congenital Heart Disease in the General Population From 2000 to 2010. Circulation 2014, 130, 749–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacha, E.A.; Cooper, D.; Thiagarajan, R.; Franklin, R.C.; Krogmann, O.; Deal, B.; Mavroudis, C.; Shukla, A.; Yeh, T.; Ba-rach, P.; et al. Cardiac complications associated with the treatment of patients with congenital cardiac disease: Consensus definitions from the Multi-Societal Database Committee for Pediatric and Congenital Heart Disease. Cardiol. Young 2008, 18 (Suppl. 2), 196–201. [Google Scholar] [CrossRef] [PubMed]
- Pierpont, M.E.; Brueckner, M.; Chung, W.K.; Garg, V.; Lacro, R.V.; McGuire, A.L.; Mital, S.; Priest, J.R.; Pu, W.T.; Roberts, A.; et al. Genetic Basis for Congenital Heart Disease: Revisited: A Scientific Statement From the American Heart Association. Circulation 2018, 138, e653–e711. [Google Scholar] [CrossRef] [PubMed]
- Pierpont, M.E.; Basson, C.T.; Benson, D.W.; Gelb, B.D.; Giglia, T.M.; Goldmuntz, E.; McGee, G.; Sable, C.A.; Srivastava, D.; Webb, C.L. Genetic Basis for Congenital Heart Defects: Current Knowledge—A scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young. Circulation 2007, 115, 3015–3038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, S.; Brueckner, M. Genetics and Genomics of Congenital Heart Disease. Circ. Res. 2017, 120, 923–940. [Google Scholar] [CrossRef]
- Mitchell, M.E.; Sander, T.L.; Klinkner, D.B.; Tomita-Mitchell, A. The Molecular Basis of Congenital Heart Disease. Semin. Thorac. Cardiovasc. Surg. 2007, 19, 228–237. [Google Scholar] [CrossRef]
- Cowan, J.R.; Ware, S.M. Genetics and Genetic Testing in Congenital Heart Disease. Clin. Perinatol. 2015, 42, 373–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuciene, R.; Dulskiene, V. Selected environmental risk factors and congenital heart defects. Medicina 2008, 44, 827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meberg, A.; Hals, J.; Thaulow, E. Congenital heart defects—Chromosomal anomalies, syndromes and extracardiac malformations. Acta Paediatr. 2007, 96, 1142–1145. [Google Scholar] [CrossRef]
- Van Der Bom, T.; Zomer, A.C.; Zwinderman, A.H.; Meijboom, F.J.; Bouma, B.J.; Mulder, B.J.M. The changing epidemiology of congenital heart disease. Nat. Rev. Cardiol. 2010, 8, 50–60. [Google Scholar] [CrossRef]
- Brown, C.B.; Wenning, J.M.; Lu, M.M.; Epstein, D.J.; Meyers, E.N.; Epstein, J.A. Cremediated excision of Fgf8 in the Tbx1 expression domain reveals a critical role for Fgf8 in cardiovascular development in the mouse. Dev. Biol. 2004, 267, 190–202. [Google Scholar] [CrossRef] [Green Version]
- Farr, G.H.; Imani, K.; Pouv, D.; Maves, L. Functional testing of a human PBX3 variant in zebrafish reveals a potential modifier role in congenital heart defects. Dis. Model. Mech. 2018, 11, dmm035972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Morton, S.; DePalma, S.R.; Zeng, X.; Qi, H.; Chang, W.; Sierant, M.C.; et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet. 2017, 49, 1593–1601. [Google Scholar] [CrossRef] [Green Version]
- Millar, R.; Barnhart, W.S. Dr. Maude Abbott’s Atlas of Congenital Cardiac Disease. Can. Med. Assoc. J. 1936, 34, 194–195. [Google Scholar]
- Witman, N.; Zhou, C.; Beverborg, N.G.; Sahara, M.; Chien, K.R. Cardiac progenitors and paracrine mediators in cardiogenesis and heart regeneration. Semin. Cell Dev. Biol. 2020, 100, 29–51. [Google Scholar] [CrossRef] [PubMed]
- Devine, W.P.; Wythe, J.D.; George, M.; Koshiba-Takeuchi, K.; Bruneau, B.G. Early patterning and specification of cardiac progenitors in gastrulating mesoderm. eLife 2014, 3, e03848. [Google Scholar] [CrossRef]
- Takeuchi, J.K.; Bruneau, B.G. Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature 2009, 459, 708–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Vliet, P.; Wu, S.M.; Zaffran, S.; Puceat, M. Early cardiac development: A view from stem cells to embryos. Cardiovasc. Res. 2012, 96, 352–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregoire, S.; Karra, R.; Passer, D.; Deutsch, M.-A.; Krane, M.; Feistritzer, R.; Sturzu, A.; Domian, I.; Saga, Y.; Wu, S.M. Essential and unexpected role of Yin Yang 1 to promote mesodermal cardiac differentiation. Circ. Res. 2013, 112, 900–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, R.P. Patterning the vertebrate heart. Nat. Rev. Genet. 2002, 3, 544–556. [Google Scholar] [CrossRef]
- Moorman, A.F.M.; Christoffels, V.M.; Anderson, R.H.; Hoff, M.J.B.V.D. The heart-forming fields: One or multiple? Philos. Trans. R. Soc. B Biol. Sci. 2007, 362, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sizarov, A.; Ya, J.; De Boer, B.A.; Lamers, W.H.; Christoffels, V.M.; Moorman, A.F.M. Formation of the Building Plan of the Human Heart: Morphogenesis, growth, and differentiation. Circulation 2011, 123, 1125–1135. [Google Scholar] [CrossRef] [Green Version]
- Van Den Hoff, M.J.B.; Kruithof, B.P.T.; Moorman, A.F.M. Making more heart muscle. BioEssays 2004, 26, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.F.; Schumacher, C.A.; de Boer, P.A.; Hagoorta, J.; Bezstarostib, K.; Hoff, M.J.V.D.; Wagenaar, G.T.; Lamers, J.M.; Wuytackc, F.; Christoffels, V.M.; et al. Presence of Functional Sarcoplasmic Reticulum in the Developing Heart and Its Confinement to Chamber Myocardium. Dev. Biol. 2000, 223, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Moorman, A.F.M.; Christoffels, V.M. Cardiac Chamber Formation: Development, Genes, and Evolution. Physiol. Rev. 2003, 83, 1223–1267. [Google Scholar] [CrossRef] [PubMed]
- Tyser, R.C.; Miranda, A.M.; Chen, C.-M.; Davidson, S.M.; Srinivas, S.; Riley, P.R. Calcium handling precedes cardiac differentiation to initiate the first heartbeat. eLife 2016, 5, e17113. [Google Scholar] [CrossRef]
- Bayraktar, M.; Männer, J. Cardiac looping may be driven by compressive loads resulting from unequal growth of the heart and pericardial cavity. Observations on a physical simulation model. Front. Physiol. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Männer, J. The anatomy of cardiac looping: A step towards the understanding of the morphogenesis of several forms of congenital cardiac malformations. Clin. Anat. 2008, 22, 21–35. [Google Scholar] [CrossRef]
- De Boer, B.A.; Berg, G.V.D.; De Boer, P.A.; Moorman, A.F.; Ruijter, J.M. Growth of the developing mouse heart: An interactive qualitative and quantitative 3D atlas. Dev. Biol. 2012, 368, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soufan, A.T.; Berg, G.V.D.; Ruijter, J.M.; De Boer, P.A.J.; Hoff, M.J.B.V.D.; Moorman, A.F.M. Regionalized Sequence of Myocardial Cell Growth and Proliferation Characterizes Early Chamber Formation. Circ. Res. 2006, 99, 545–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norden, J.; Kispert, A. Wnt/Ctnnb1 Signaling and the Mesenchymal Precursor Pools of the Heart. Trends Cardiovasc. Medicine 2012, 22, 118–122. [Google Scholar] [CrossRef]
- Ruiz-Villalba, A.; Hoppler, S.; Hoff, M.J.V.D. Wnt signaling in the heart fields: Variations on a common theme. Dev. Dyn. 2016, 245, 294–306. [Google Scholar] [CrossRef]
- Buckingham, M.; Meilhac, S.M.; Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef]
- Vincent, S.D.; Buckingham, M.E. How to make a heart. The origin and regulation of cardiac progenitor cells. Curr. Top. Dev. Biol. 2010, 90, 1–41. [Google Scholar]
- Schultheiss, T.M.; Burch, J.B.; Lassar, A.B. A role for bone morphogenetic proteins in the induction of cardiac myogenesis. Genes Dev. 1997, 11, 451–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reifers, F.; Walsh, E.C.; Léger, S.; Stainier, D.Y.; Brand, M. Induction and differentiation of the zebrafish heart requires fibroblast growth factor 8 (fgf8/acerebellar). Development 2000, 127, 225–235. [Google Scholar] [PubMed]
- Pandur, P.; Läsche, M.; Eisenberg, L.M.; Kühl, M. Wnt-11 activation of a non-canonical Wnt signalling pathway is required for cardiogenesis. Nature 2002, 418, 636–641. [Google Scholar] [CrossRef]
- Cai, C.-L.; Liang, X.; Shi, Y.; Chu, P.-H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 Identifies a Cardiac Progenitor Population that Proliferates Prior to Differentiation and Contributes a Majority of Cells to the Heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef] [Green Version]
- Ilagan, R.; Abu-Issa, R.; Brown, D.; Yang, Y.-P.; Jiao, K.; Schwartz, R.J.; Klingensmith, J.; Meyers, E.N. Fgf8 is required for anterior heart field development. Development 2006, 133, 2435–2445. [Google Scholar] [CrossRef] [Green Version]
- Dyer, L.A.; Kirby, M.L. Sonic hedgehog maintains proliferation in secondary heart field progenitors and is required for normal arterial pole formation. Dev. Biol. 2009, 330, 305–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, E.D.; Wang, Z.; Lepore, J.J.; Lu, M.M.; Taketo, M.M.; Epstein, D.J.; Morrisey, E.E. Wnt/β-catenin signaling promotes expansion of Isl-1–positive cardiac progenitor cells through regulation of FGF signaling. J. Clin. Investig. 2007, 117, 1794–1804. [Google Scholar] [CrossRef] [Green Version]
- De Bono, C.; Thellier, C.; Bertrand, N.; Sturny, R.; Jullian, E.; Cortes, C.; Stefanovic, S.; Zaffran, S.; Théveniau-Ruissy, M.; Kelly, R.G. T-box genes and retinoic acid signaling regulate the segregation of arterial and venous pole progenitor cells in the murine second heart field. Hum. Mol. Genet. 2018, 27, 3747–3760. [Google Scholar] [CrossRef] [PubMed]
- Rana, M.S.; Théveniau-Ruissy, M.; De Bono, C.; Mesbah, K.; Francou, A.; Rammah, M.; Domínguez, J.N.; Roux, M.; Laforest, B.; Anderson, R.H.; et al. Tbx1 Coordinates Addition of Posterior Second Heart Field Progenitor Cells to the Arterial and Venous Poles of the Heart. Circ. Res. 2014, 115, 790–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutson, M.R.; Zeng, X.L.; Kim, A.J.; Antoon, E.; Harward, S.; Kirby, M.L. Arterial pole progenitors interpret opposing FGF/BMP signals to proliferate or differentiate. Development 2010, 137, 3001–3011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.; Li, D.; Gupta, M.; Manderfield, L.J.; Ifkovits, J.L.; Wang, Q.; Liu, F.; Liu, Y.; Poleshko, A.; Padmanabhan, A.; et al. Integration of Bmp and Wnt signaling by Hopx specifies commitment of cardiomyoblasts. Science 2015, 348, aaa6071. [Google Scholar] [CrossRef] [Green Version]
- Klaus, A.; Müller, M.; Schulz, H.; Saga, Y.; Martin, J.F.; Birchmeier, W. Wnt/β-catenin and Bmp signals control distinct sets of transcription factors in cardiac progenitor cells. Proc. Natl. Acad. Sci. USA 2012, 109, 10921–10926. [Google Scholar] [CrossRef] [Green Version]
- Lien, C.-L.; McAnally, J.; Richardson, J.A.; Olson, E.N. Cardiac-Specific Activity of an Nkx2–5 Enhancer Requires an Evolutionarily Conserved Smad Binding Site. Dev. Biol. 2002, 244, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Paige, S.L.; Plonowska, K.; Xu, A.; Wu, S.M. Molecular regulation of cardiomyocyte differentiation. Circ. Res. 2015, 116, 341–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Cai, C.-L.; Lin, L.; Qyang, Y.; Chung, C.; Monteiro, R.M.; Mummery, C.L.; Fishman, G.I.; Cogen, A.; Evans, S. Isl1Cre reveals a common Bmp pathway in heart and limb development. Development 2006, 133, 1575–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, C.; Arnold, J.; Hsiao, E.C.; Taketo, M.M.; Conklin, B.R.; Srivastava, D. Canonical Wnt signaling is a positive regulator of mammalian cardiac progenitors. Proc. Natl. Acad. Sci. USA 2007, 104, 10894–10899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, C.; Qian, L.; Cheng, P.; Nigam, V.; Arnold, J.; Srivastava, D. A regulatory pathway involving Notch1/β-catenin/Isl1 determines cardiac progenitor cell fate. Nature 2009, 11, 951–957. [Google Scholar] [CrossRef] [Green Version]
- Klaus, A.; Saga, Y.; Taketo, M.M.; Tzahor, E.; Birchmeier, W. Distinct roles of Wnt/beta-catenin and Bmp signaling during early cardiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 18531–18536. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, C.A.; Eisenberg, L.M. WNT11 promotes cardiac tissue formation of early mesoderm. Dev. Dyn. 1999. [Google Scholar] [CrossRef]
- Cohen, E.D.; Miller, M.F.; Wang, Z.; Moon, R.T.; Morrisey, E.E. Wnt5a and Wnt11 are essential for second heart field progenitor development. Development 2012, 139, 1931–1940. [Google Scholar] [CrossRef] [Green Version]
- Yutzey, K.E.; Rhee, J.T.; Bader, D. Expression of the atrial-specific myosin heavy chain AMHC1 and the establishment of anteroposterior polarity in the developing chicken heart. Development 1994, 120, 871–883. [Google Scholar]
- Mommersteeg, M.T.M.; Hoogaars, W.M.H.; Prall, O.W.J.; Vries, C.D.G.-D.; Wiese, C.; Clout, D.E.W.; Papaioannou, V.E.; Brown, N.A.; Harvey, R.P.; Moorman, A.F.M.; et al. Molecular Pathway for the Localized Formation of the Sinoatrial Node. Circ. Res. 2007, 100, 354–362. [Google Scholar] [CrossRef] [Green Version]
- Goddeeris, M.M.; Rho, S.; Petiet, A.; Davenport, C.L.; Johnson, G.A.; Meyers, E.N.; Klingensmith, J. Intracardiac septation requires hedgehog-dependent cellular contributions from outside the heart. Development 2008, 135, 1887–1895. [Google Scholar] [CrossRef] [Green Version]
- Snarr, B.S.; O’Neal, J.L.; Chintalapudi, M.R.; Wirrig, E.E.; Phelps, A.L.; Kubalak, S.W.; Wessels, A. Isl1 Expression at the Venous Pole Identifies a Novel Role for the Second Heart Field in Cardiac Development. Circ. Res. 2007, 101, 971–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harh, J.Y.; Paul, M.H. Experimental cardiac morphogenesis. I. Development of the ventricular septum in the chick. J. Embryol. Exp. Morphol. 1975, 33, 13–28. [Google Scholar] [PubMed]
- Lopez, L.; Houyel, L.; Colan, S.D.; Anderson, R.H.; Béland, M.J.; Aiello, V.D.; Bailliard, F.; Cohen, M.S.; Jacobs, J.P.; Kurosawa, H.; et al. Classification of Ventricular Septal Defects for the Eleventh Iteration of the International Classification of Diseases—Striving for Consensus: A Report From the International Society for Nomenclature of Paediatric and Congenital Heart Disease. Ann. Thorac. Surg. 2018, 106, 1578–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sizarov, A.; Lamers, W.H.; Mohun, T.J.; Brown, N.A.; Anderson, R.H.; Moorman, A.F.M. Three-dimensional and molecular analysis of the arterial pole of the developing human heart. J. Anat. 2012, 220, 336–349. [Google Scholar] [CrossRef]
- Rana, M.S.; Sizarov, A.; Christoffels, V.M.; Moorman, A.F.M. Development of the human aortic arch system captured in an interactive three-dimensional reference model. Am. J. Med. Genet. Part A 2013, 164, 1372–1383. [Google Scholar] [CrossRef] [PubMed]
- Rana, M.S.; Horsten, N.C.; Tesink-Taekema, S.; Lamers, W.H.; Moorman, A.F.; Hoff, M.J.V.D. Trabeculated Right Ventricular Free Wall in the Chicken Heart Forms by Ventricularization of the Myocardium Initially Forming the Outflow Tract. Circ. Res. 2007, 100, 1000–1007. [Google Scholar] [CrossRef] [Green Version]
- Webb, S.; Qayyum, S.R.; Anderson, R.H.; Lamers, W.H.; Richardson, M.K. Septation and separation within the outflow tract of the developing heart. J. Anat. 2003, 202, 327–342. [Google Scholar] [CrossRef]
- Kelly, R.G.; Buckingham, M.E. The anterior heart-forming field: Voyage to the arterial pole of the heart. Trends Genet. 2002, 18, 210–216. [Google Scholar] [CrossRef]
- Jiang, X.; Rowitch, D.H.; Soriano, P.; McMahon, A.P.; Sucov, H.M. Fate of the mammalian cardiac neural crest. Development 2000, 127, 1607–1616. [Google Scholar] [PubMed]
- Stoller, J.Z.; Epstein, J.A. Cardiac neural crest. Semin. Cell Dev. Biol. 2005, 16, 704–715. [Google Scholar] [CrossRef]
- Ya, J.; Hoff, M.J.B.V.D.; De Boer, P.A.J.; Tesink-Taekema, S.; Franco, D.; Moorman, A.F.M.; Lamers, W.H. Normal Development of the Outflow Tract in the Rat. Circ. Res. 1998, 82, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.H.; Chaudhry, B.; Mohun, T.J.; Bamforth, S.D.; Hoyland, D.; Phillips, H.M.; Webb, S.; Moorman, A.F.; Brown, N.A.; Henderson, D.J. Normal and abnormal development of the intrapericardial arterial trunks in humans and mice. Cardiovasc. Res. 2012, 95, 108–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, N.; Akimoto, N.; Hidaka, N.; Shoji, S.; Sumida, H. Formal genesis of the outflow tracts of the heart revisited: Previous works in the light of recent observations. Congenit. Anom. 2010, 50, 141–158. [Google Scholar] [CrossRef]
- Hutson, M.R.; Kirby, M.L. Neural crest and cardiovascular development: A 20-year perspective. Birth Defects Res. Part C Embryo Today Rev. 2003, 69, 2–13. [Google Scholar] [CrossRef]
- Waddington, C.H. Genetic Assimilation of the Bithorax Phenotype. Evolution 1956, 10. [Google Scholar] [CrossRef]
- Feinberg, A.P. The Key Role of Epigenetics in Human Disease Prevention and Mitigation. N. Engl. J. Med. 2018, 378, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Yu, Z.-B.; Chen, X.-H.; Ji, C.-B.; Qian, L.-M.; Han, S.-P. DNA hypermethylation of the NOX5 gene in fetal ventricular septal defect. Exp. Ther. Med. 2011, 2, 1011–1015. [Google Scholar] [CrossRef]
- Zhu, C.; Yu, Z.B.; Chen, X.H.; Pan, Y.; Dong, X.Y.; Qian, L.M.; Han, S.-P. Screening for differential methylation status in fetal myocardial tissue samples with ventricular septal defects by promoter methylation microarrays. Mol. Med. Rep. 2010, 4, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Sheng, W.; Wang, H.; Ma, X.; Qian, Y.; Zhang, P.; Wu, Y.; Zheng, F.; Chen, L.; Huang, G.; Ma, D. LINE-1 methylation status and its association with tetralogy of fallot in infants. BMC Med. Genomics 2012, 5, 20. [Google Scholar] [CrossRef] [Green Version]
- Sheng, W.; Qian, Y.; Wang, H.; Ma, X.; Zhang, P.; Chen, L.; Ma, D.; Huang, G. Association between mRNA levels of DNMT1, DNMT3A, DNMT3B, MBD2 and LINE-1 methylation status in infants with tetralogy of Fallot. Int. J. Mol. Med. 2013, 32, 694–702. [Google Scholar] [CrossRef]
- Sheng, W.; Qian, Y.; Wang, H.; Ma, X.; Zhang, P.; Diao, L.; An, Q.; Chen, L.; Ma, D.; Huang, G. DNA methylation status of NKX2-5, GATA4 and HAND1in patients with tetralogy of fallot. BMC Med. Genomics 2013, 6, 46. [Google Scholar] [CrossRef] [Green Version]
- Sheng, W.; Qian, Y.; Zhang, P.; Wu, Y.; Wang, H.; Ma, X.; Chen, L.; Ma, D.; Huang, G. Association of promoter methylation statuses of congenital heart defect candidate genes with Tetralogy of Fallot. J. Transl. Med. 2014, 12, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra-Juhé, C.; Cuscó, I.; Homs, A.; Flores, R.; Torán, N.; Pérez-Jurado, L.A. DNA methylation abnormalities in congenital heart disease. Epigenetics 2015, 10, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Grunert, M.; Dorn, C.; Cui, H.; Dunkel, I.; Schulz, K.; Schoenhals, S.; Sun, W.; Berger, F.; Chen, W.; Sperling, S.R. Comparative DNA methylation and gene expression analysis identifies novel genes for structural congenital heart diseases. Cardiovasc. Res. 2016, 112, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; Chen, L.; Wang, H.; Ma, X.; Ma, D.; Huang, G. CpG island shore methylation of ZFPM2 is identified in tetralogy of fallot samples. Pediatr. Res. 2016, 80, 151–158. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.-J.; Zhang, G.-F.; Zhang, R.-P. High CpG island methylation of p16 gene and loss of p16 protein expression associate with the development and progression of tetralogy of Fallot. J. Genet. 2016, 95, 831–837. [Google Scholar] [CrossRef]
- Qian, Y.; Xiao, D.; Guo, X.; Chen, H.; Hao, L.; Ma, X.; Huang, G.; Ma, D.; Wang, H. Hypomethylation and decreased expression of BRG1 in the myocardium of patients with congenital heart disease. Birth Defects Res. 2017, 109, 1183–1195. [Google Scholar] [CrossRef]
- Asim, A.; Agarwal, S.; Panigrahi, I.; Saiyed, N.; Bakshi, S. MTHFR promoter hypermethylation may lead to congenital heart defects in Down syndrome. Intractable Rare Dis. Res. 2017, 6, 295–298. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Kong, Q.; Li, Z.; Xu, M.; Cai, Z.; Zhao, C. Association between the promoter methylation of the TBX20 gene and tetralogy of fallot. Scand. Cardiovasc. J. 2018, 52, 287–291. [Google Scholar] [CrossRef]
- Gong, J.; Sheng, W.; Ma, D.; Huang, G.; Liu, F. DNA methylation status of TBX20 in patients with tetralogy of Fallot. BMC Med. Genomics 2019, 12, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, G.; Zhang, C.; Ling, T.; Liu, R.; Zong, L.; Guan, Y.; Huang, X.; Sun, L.; Zhang, L.; Li, C.; et al. Genome and epigenome analysis of monozygotic twins discordant for congenital heart disease. BMC Genomics 2018, 19, 1–13. [Google Scholar] [CrossRef]
- Dobosz, A.; Grabowska, A.; Bik-Multanowski, M. Hypermethylation of NRG1 gene correlates with the presence of heart defects in Down’s syndrome. J. Genet. 2019, 98, 110. [Google Scholar] [CrossRef] [PubMed]
- Nimura, K.; Ura, K.; Shiratori, H.; Ikawa, M.; Okabe, M.; Schwartz, R.J.; Kaneda, Y. A histone H3 lysine 36 trimethyl-transferase links Nkx2-5 to Wolf–Hirschhorn syndrome. Nature 2009, 460, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Von Elten, K.; Sawyer, T.; Lentz-Kapua, S.; Kanis, A.; Studer, M. A Case of Wolf-Hirschhorn Syndrome and Hypoplastic Left Heart Syndrome. Pediatr. Cardiol. 2012, 34, 1244–1246. [Google Scholar] [CrossRef] [PubMed]
- Cooper, H.; Hirschhorn, K. Apparent deletion of short arms of one chromosome (4 or 5) in a child with defects of midline fusion. Mamm. Chromosom. Newsl. 1961, 4, 479–482. [Google Scholar]
- Zaidi, S.; Choi, M.; Wakimoto, H.; Ma, L.; Jiang, J.; Overton, J.D.; Romano-Adesman, A.; Bjornson, R.D.; Breitbart, R.E.; Brown, K.K.; et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013, 498, 220–223. [Google Scholar] [CrossRef] [Green Version]
- Su, D.; Li, Q.; Guan, L.; Gao, X.; Zhang, H.; Dandan, E.; Zhang, L.; Ma, X. Down-regulation of EBAF in the heart with ventricular septal defects and its regulation by histone acetyltransferase p300 and transcription factors smad2 and cited2. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2013, 1832, 2145–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robson, A.; Makova, S.Z.; Barish, S.; Zaidi, S.; Mehta, S.; Drozd, J.; Jin, S.C.; Gelb, B.D.; Seidman, C.E.; Chung, W.K.; et al. Histone H2B monoubiquitination regulates heart development via epigenetic control of cilia motility. Proc. Natl. Acad. Sci. USA 2019, 116, 14049–14054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Chung, J.H.; Wang, T.; McDonald-McGinn, D.M.; Kates, W.R.; Hawuła, W.; Coleman, K.; Zackai, E.; Emanuel, B.S.; Morrow, B.E. Histone Modifier Genes Alter Conotruncal Heart Phenotypes in 22q11.2 Deletion Syndrome. Am. J. Hum. Genet. 2015, 97, 869–877. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.-S.; Wang, J.-Z.; Shi, S.; Han, Y.; Zhang, Y.; Zhi, J.-X.; Xu, C.; Li, F.-F.; Wang, G.-Y.; Liu, S.-L. Identification of epigenetic factor KAT2B gene variants for possible roles in congenital heart diseases. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Subrahmanyan, L.; Smith, E.; Yu, X.; Zaidi, S.; Choi, M.; Mane, S.; Nelson-Williams, C.; Bahjati, M.; Kazemi, M.; et al. Mutations in the Histone Modifier PRDM6 Are Associated with Isolated Nonsyndromic Patent Ductus Arteriosus. Am. J. Hum. Genet. 2016, 98, 1082–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- León, L.E.; Benavides, F.; Espinoza, K.; Vial, C.; Alvarez, P.; Palomares, M.; Lay-Son, G.; Miranda, M.; Repetto, G.M. Partial microduplication in the histone acetyltransferase complex member KANSL1 is associated with congenital heart de-fects in 22q11.2 microdeletion syndrome patients. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef]
- Xu, J.; Hu, Z.; Xu, Z.; Gu, H.; Yi, L.; Cao, H.; Chen, J.; Tian, T.; Liang, J.; Lin, Y.; et al. Functional variant in microRNA-196a2 contributes to the susceptibility of congenital heart disease in a Chinese population. Hum. Mutat. 2009, 30, 1231–1236. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, Y.; Ma, X.-J.; Wang, H.-J.; Zhang, J.; Luo, X.; Chen, W.; Wu, Y.; Meng, Y.; Yuan, Y.; et al. Roles of miR-1-1 and miR-181c in ventricular septal defects. Int. J. Cardiol. 2013, 168, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ma, X.-J.; Wang, H.-J.; Li, W.-C.; Chen, L.; Ma, D.; Huang, G.-Y. Expression of Cx43-related microRNAs in patients with tetralogy of Fallot. World J. Pediatr. 2013, 10, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ji, L.; Liu, L.; Liu, Y.; Hou, H.; Yu, K.; Sun, Q.; Zhao, Z. Characterization of Circulating MicroRNA Expression in Patients with a Ventricular Septal Defect. PLoS ONE 2014, 9, e106318. [Google Scholar] [CrossRef]
- Huang, J.; Li, X.; Li, H.; Su, Z.; Wang, J.; Zhang, H. Down-regulation of microRNA-184 contributes to the development of cyanotic congenital heart diseases. Int. J. Clin. Exp. Pathol. 2015, 8, 14221–14227. [Google Scholar]
- Wang, Y.; Du, X.; Zhou, Z.; Jiang, J.; Zhang, Z.; Ye, L.; Hong, H. A gain-of-function ACTC1 3′UTR mutation that introduces a miR-139-5p target site may be associated with a dominant familial atrial septal defect. Sci. Rep. 2016, 6, 25404. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Li, Z.; Song, X.; Liu, L.; Su, G.; Cui, Y. Bioinformatic Analysis of Genes and MicroRNAs Associated With Atrioventricular Septal Defect in Down Syndrome Patients. Int. Heart J. 2016, 57, 490–495. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Yang, L.; Luo, H.; Tan, F.; Ma, X.; Lu, C. A Rare Rs139365823 Polymorphism in Pre-miR-138 Is Associated with Risk of Congenital Heart Disease in a Chinese Population. DNA Cell Biol. 2018, 37, 109–116. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Takai, D.; Jones, P.A. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. USA 2002, 99, 3740–3745. [Google Scholar] [CrossRef] [Green Version]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for De Novo Methylation and Mammalian Development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilsbach, R.; Preissl, S.; Grüning, B.A.; Schnick, T.; Burger, L.; Benes, V.; Würch, A.; Bönisch, U.; Günther, S.; Backofen, R.; et al. Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat. Commun. 2014, 5, 5288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crider, K.S.; Yang, T.P.; Berry, R.J.; Bailey, L.B. Folate and DNA Methylation: A Review of Molecular Mechanisms and the Evidence for Folate’s Role. Adv. Nutr. 2012, 3, 21–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locke, A.E.; Dooley, K.J.; Tinker, S.W.; Cheong, S.Y.; Feingold, E.; Allen, E.G.; Freeman, S.B.; Torfs, C.P.; Cua, C.L.; Epstein, M.P.; et al. Variation in folate pathway genes contributes to risk of congenital heart defects among individuals with Down syndrome. Genet. Epidemiology 2010, 34, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Yin, M.; Dong, L.; Zheng, J.; Zhang, H.; Liu, J.; Xu, Z. Meta Analysis of the Association between MTHFR C677T Poly-morphism and the Risk of Congenital Heart Defects. Ann. Hum. Genet. 2011, 76, 9–16. [Google Scholar] [CrossRef]
- Bean, L.J.H.; Allen, E.G.; Tinker, S.W.; Hollis, N.D.; Locke, A.E.; Druschel, C.; Hobbs, C.A.; O’Leary, L.; Romitti, P.A.; Royle, M.H.; et al. Lack of maternal folic acid supplementation is associated with heart defects in Down syndrome: A report from the National Down Syndrome Project. Birth Defects Res. Part A Clin. Mol. Teratol. 2011, 91, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Obermann-Borst, S.A.; Van Driel, L.M.J.W.; Helbing, W.A.; De Jonge, R.; Wildhagen, M.F.; Steegers, E.A.P.; Steegers-Theunissen, R.P.M. Congenital heart defects and biomarkers of methylation in children: A case-control study. Eur. J. Clin. Investig. 2010, 41, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Van Driel, L.M.J.W.; De Jonge, R.; Helbing, W.A.; Van Zelst, B.D.; Ottenkamp, J.; Steegers, E.A.P.; Steegers-Theunissen, R.P.M. Maternal Global Methylation Status and Risk of Congenital Heart Diseases. Obstet. Gynecol. 2008, 112, 277–283. [Google Scholar] [CrossRef]
- Bahado-Singh, R.O.; Zaffra, R.; Albayarak, S.; Chelliah, A.; Bolinjkar, R.; Turkoglu, O.; Radhakrishna, U. Epigenetic markers for newborn congenital heart defect (CHD). J. Matern. Neonatal Med. 2015, 29, 1–7. [Google Scholar] [CrossRef]
- Fenech, M. Cytokinesis-block micronucleus assay evolves into a “cytome” assay of chromosomal instability, mitotic dysfunction and cell death. Mutat. Res. Mol. Mech. Mutagen. 2006, 600, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M. Micronuclei and their association with sperm abnormalities, infertility, pregnancy loss, pre-eclampsia and intra-uterine growth restriction in humans. Mutagenesis 2011, 26, 63–67. [Google Scholar] [CrossRef]
- Radhakrishna, U.; Albayrak, S.; Zafra, R.; Baraa, A.; Vishweswaraiah, S.; Veerappa, A.M.; Mahishi, D.; Saiyed, N.; Mishra, N.K.; Guda, C.; et al. Placental epigenetics for evaluation of fetal congenital heart defects: Ventricular Septal Defect (VSD). PLoS ONE 2019, 14, e0200229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, Y.-A.; Arnold, R.S.; Lassegue, B.; Shi, J.; Xu, X.; Sorescu, D.; Chung, A.B.; Griendling, K.K.; Lambeth, J.D. Cell transformation by the superoxide-generating oxidase Mox1. Nature 1999, 401, 79–82. [Google Scholar] [CrossRef]
- Piao, Y.J.; Seo, Y.H.; Hong, F.; Kim, J.H.; Kim, Y.-J.; Kang, M.H.; Kim, B.S.; Jo, S.A.; Jo, I.; Jue, D.-M.; et al. Nox 2 stimulates muscle differentiation via NF-κB/iNOS pathway. Free. Radic. Biol. Med. 2005, 38, 989–1001. [Google Scholar] [CrossRef]
- Pedruzzi, E.; Guichard, C.; Ollivier, V.; Driss, F.; Fay, M.; Prunet, C.; Marie, J.-C.; Pouzet, C.; Samadi, M.; Elbim, C.; et al. NAD(P)H Oxidase Nox-4 Mediates 7-Ketocholesterol-Induced Endoplasmic Reticulum Stress and Apoptosis in Human Aortic Smooth Muscle Cells. Mol. Cell. Biol. 2004, 24, 10703–10717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijnands, K.P.; Chen, J.; Liang, L.; Verbiest, M.M.; Lin, X.; Helbing, W.A.; Groot, A.C.G.-D.; Van Der Spek, P.J.; Uitterlinden, A.G.; Steegers-Theunissen, R.P. Genome-wide methylation analysis identifies novel CpG loci for perimembranous ventricular septal defects in human. Epigenomics 2017, 9, 241–251. [Google Scholar] [CrossRef]
- Arndt, A.-K.; Schafer, S.; Drenckhahn, J.-D.; Sabeh, M.K.; Plovie, E.R.; Caliebe, A.; Klopocki, E.; Musso, G.; Werdich, A.A.; Kalwa, H.; et al. Fine Mapping of the 1p36 Deletion Syndrome Identifies Mutation of PRDM16 as a Cause of Cardiomyopathy. Am. J. Hum. Genet. 2013, 93, 67–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjork, B.C.; Turbe-Doan, A.; Prysak, M.; Herron, B.J.; Beier, D.R. Prdm16 is required for normal palatogenesis in mice. Hum. Mol. Genet. 2009, 19, 774–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazazian, H.H.J. Mobile Elements: Drivers of Genome Evolution. Science 2004, 303, 1626–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radhakrishna, U.; Vishweswaraiah, S.; Veerappa, A.M.; Zafra, R.; Albayrak, S.; Sitharam, P.H.; Saiyed, N.M.; Mishra, N.K.; Guda, C.; Bahado-Singh, R. Newborn blood DNA epigenetic variations and signaling pathway genes associated with Tetralogy of Fallot (TOF). PLoS ONE 2018, 13, e0203893. [Google Scholar] [CrossRef]
- Cai, X.; Zhang, W.; Hu, J.; Zhang, L.; Sultana, N.; Wu, B.; Cai, W.; Zhou, B.; Cai, C.-L. Tbx20 acts upstream of Wnt signaling to regulate endocardial cushion formation and valve remodeling during mouse cardiogenesis. Development 2013, 140, 3176–3187. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Nomura-Kitabayashi, A.; Cai, W.; Yan, J.; Christoffels, V.M.; Cai, C.-L. Myocardial Tbx20 regulates early atrioventricular canal formation and endocardial epithelial–mesenchymal transition via Bmp2. Dev. Biol. 2011, 360, 381–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S.; Yutzey, K.E. Tbx20 regulation of cardiac cell proliferation and lineage specialization during embryonic and fetal development in vivo. Dev. Biol. 2012, 363, 234–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, J.J.; Gelb, B.D. Genetics of congenital heart disease. Curr. Opin. Cardiol. 2016, 31, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Hirai, M.; Ono, K.; Morimoto, T.; Kawamura, T.; Wada, H.; Kita, T.; Hasegawa, K. FOG-2 Competes with GATA-4 for Transcriptional Coactivator p300 and Represses Hypertrophic Responses in Cardiac Myocytes. J. Biol. Chem. 2004, 279, 37640–37650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolstein, J.M.; Lee, D.H.; Michaud, J.; Buot, V.; Stefanchik, B.; Plotkin, M.D. INK4a knockout mice exhibit increased fibrosis under normal conditions and in response to unilateral ureteral obstruction. Am. J. Physiol. Physiol. 2010, 299, F1486–F1495. [Google Scholar] [CrossRef] [Green Version]
- An, S.; Chen, Y.; Gao, C.; Qin, B.; Du, X.; Meng, F.; Qi, Y. Inactivation of INK4a and ARF induces myocardial proliferation and improves cardiac repair following ischemia-reperfusion. Mol. Med. Rep. 2015, 12, 5911–5916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Morentin, L.; Martínez, L.; Piloto, S.; Yang, H.; Schon, E.A.; Garesse, R.; Bodmer, R.; Ocorr, K.; Cervera, M.; Arredondo, J.J. Cardiac deficiency of single cytochrome oxidase assembly factor scox induces p53-dependent apoptosis in a Drosophila cardiomyopathy model. Hum. Mol. Genet. 2015, 24, 3608–3622. [Google Scholar] [CrossRef] [Green Version]
- Bahado-Singh, R.O.; Vishweswaraiah, S.; Aydas, B.; Yilmaz, A.; Saiyed, N.M.; Mishra, N.K.; Guda, C.; Radhakrishna, U. Precision cardiovascular medicine: Artificial intelligence and epigenetics for the pathogenesis and prediction of coarctation in neonates. J. Matern. Neonatal Med. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishna, U.; Albayrak, S.; Alpay-Savasan, Z.; Zeb, A.; Turkoglu, O.; Sobolewski, P.; Bahado-Singh, R.O. Genome-Wide DNA Methylation Analysis and Epigenetic Variations Associated with Congenital Aortic Valve Stenosis (AVS). PLoS ONE 2016, 11, e0154010. [Google Scholar] [CrossRef]
- Caramori, M.L.; Kim, Y.; Moore, J.H.; Rich, S.S.; Mychaleckyj, J.C.; Kikyo, N.; Mauer, M. Gene Expression Differences in Skin Fibroblasts in Identical Twins Discordant for Type 1 Diabetes. Diabetes 2012, 61, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Yuan, W.; Xia, Y.; Bell, C.G.; Yet, I.; Ferreira, T.; Ward, K.J.; Gao, F.; Loomis, A.K.; Hyde, C.L.; Wu, H.; et al. An integrated epigenomic analysis for type 2 diabetes susceptibility loci in monozygotic twins. Nat. Commun. 2014, 5, 5719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.C.Y.; Meaburn, E.L.; Ronald, A.R.; Price, T.S.; Jeffries, A.R.; Schalkwyk, L.C.; Plomin, R.; Mill, J.G. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol. Psychiatry 2013, 19, 495–503. [Google Scholar] [CrossRef]
- Arora, M.; Reichenberg, A.; Willfors, C.; Austin, C.; Gennings, C.; Berggren, S.; Lichtenstein, P.; Anckarsäter, H.; Tammimies, K.; Bölte, S. Fetal and postnatal metal dysregulation in autism. Nat. Commun. 2017, 8, 15493. [Google Scholar] [CrossRef]
- Castillo-Fernandez, J.E.; Spector, T.D.; Bell, J.T. Epigenetics of discordant monozygotic twins: Implications for disease. Genome Med. 2014, 6, 60. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. From The Cover: Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [Green Version]
- Al Turki, S.; Manickaraj, A.K.; Mercer, C.L.; Gerety, S.S.; Hitz, M.-P.; Lindsay, S.; D’Alessandro, L.C.; Swaminathan, G.J.; Bentham, J.; Arndt, A.-K.; et al. Rare Variants in NR2F2 Cause Congenital Heart Defects in Humans. Am. J. Hum. Genet. 2014, 94, 574–585. [Google Scholar] [CrossRef] [Green Version]
- Lei, L.; Lin, H.; Zhong, S.; Zhang, Z.; Chen, J.; Yu, X.; Liu, X.; Zhang, C.; Nie, Z.; Zhuang, J. DNA methyltransferase 1 rs16999593 genetic polymorphism decreases risk in patients with transposition of great arteries. Gene 2017, 615, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Bahado-Singh, R.; Vishweswaraiah, S.; Mishra, N.K.; Guda, C.; Radhakrishna, U. Placental DNA methylation changes in detection of tetralogy of Fallot. Ultrasound Obstet. Gynecology 2019, 55, 768–775. [Google Scholar] [CrossRef]
- Davis, C.A.; Haberland, M.; Arnold, M.A.; Sutherland, L.B.; McDonald, O.G.; Richardson, J.A.; Childs, G.; Harris, S.; Owens, G.K.; Olson, E.N. PRISM/PRDM6, a Transcriptional Repressor That Promotes the Proliferative Gene Program in Smooth Muscle Cells. Mol. Cell. Biol. 2006, 26, 2626–2636. [Google Scholar] [CrossRef] [Green Version]
- Juan, H.; Hamada, H. Roles of nodal-lefty regulatory loops in embryonic patterning of vertebrates. Genes Cells 2001, 6, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Weninger, W.J.; Ghislain, J.; Desmarquet-Trin-Dinh, C.; Gilardi-Hebenstreit, P.; Charnay, P.; Frain, M. Cited2 is required both for heart morphogenesis and establishment of the left-right axis in mouse development. Development 2005, 132, 1337–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bamforth, S.D.; Bragança, J.; Farthing, C.R.; Schneider, J.E.; Broadbent, C.; Michell, A.C.; Clarke, K.; Neubauer, S.; Norris, D.; Brown, N.A.; et al. Cited2 controls left-right patterning and heart development through a Nodal-Pitx2c pathway. Nat. Genet. 2004, 36, 1189–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordovás, J.M.; Smith, C.E. Epigenetics and cardiovascular disease. Nat. Rev. Cardiol. 2010, 7, 510–519. [Google Scholar] [CrossRef] [Green Version]
- Bartke, T.; Kouzarides, T. Decoding the chromatin modification landscape. Cell Cycle 2011, 10, 182. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Jin, J.; Swanson, S.K.; Cole, M.D.; Choi, S.H.; Florens, L.; Washburn, M.P.; Conaway, J.W.; Conaway, R.C. Subunit Composition and Substrate Specificity of a MOF-containing Histone Acetyltransferase Distinct from the Male-specific Lethal (MSL) Complex. J. Biol. Chem. 2010, 285, 4268–4272. [Google Scholar] [CrossRef] [Green Version]
- Dias, J.; Van Nguyen, N.; Georgiev, P.; Gaub, A.; Brettschneider, J.; Cusack, S.; Kadlec, J.; Akhtar, A. Structural analysis of the KANSL1/WDR5/KANSL2 complex reveals that WDR5 is required for efficient assembly and chromatin targeting of the NSL complex. Genes Dev. 2014, 28, 929–942. [Google Scholar] [CrossRef] [Green Version]
- Hacıhamdioğlu, B.; Hacihamdioglu, D.O.; Delil, K. 22q11 deletion syndrome: Current perspective. Appl. Clin. Genet. 2015, 8, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.; Veyver, I.B.V.D. 22q11.2 Deletion Syndrome. In Obstetric Imaging: Fetal Diagnosis and Care, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 621–626. [Google Scholar] [CrossRef]
- Unolt, M.; Versacci, P.; Anaclerio, S.; Lambiase, C.; Calcagni, G.; Trezzi, M.; Carotti, A.; Crowley, T.B.; Zackai, E.H.; Goldmuntz, E.; et al. Congenital heart diseases and cardiovascular abnormalities in 22q11.2 deletion syndrome: From well-established knowledge to new frontiers. Am. J. Med. Genet. Part A 2018, 176, 2087–2098. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Guermah, M.; McGinty, R.K.; Lee, J.-S.; Tang, Z.; Milne, T.A.; Shilatifard, A.; Muir, T.W.; Roeder, R.G. RAD6-Mediated Transcription-Coupled H2B Ubiquitylation Directly Stimulates H3K4 Methylation in Human Cells. Cell 2009, 137, 459–471. [Google Scholar] [CrossRef] [Green Version]
- Han, P.; Chang, C.-P.; Yang, J. Chromatin Remodeling in Cardiovascular Development and Physiology. Circ. Res. 2011, 108, 378–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stankunas, K.; Chang, C.-P.; Tsun, Z.-Y.; Chen, H.; Lee, N.V.; Wu, J.I.; Shang, C.; Bayle, J.H.; Shou, W.; Iruela-Arispe, M.L. Endocardial Brg1 Represses ADAMTS1 to Maintain the Microenvironment for Myocardial Morphogenesis. Dev. Cell 2008, 14, 298–311. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-P.; Yang, J.; Han, P.; Cheng, H.-L.; Shang, C.; Ashley, E.; Zhou, B. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature 2010, 466, 62–67. [Google Scholar] [CrossRef]
- Takeuchi, J.K.; Lou, X.; Alexander, J.M.; Sugizaki, H.; Delgado-Olguín, P.; Holloway, A.K.; Mori, A.D.; Wylie, J.N.; Mun-son, C.; Zhu, Y.; et al. Chromatin remodelling complex dosage modulates transcription factor function in heart development. Nat. Commun. 2011, 2, 187. [Google Scholar] [CrossRef] [Green Version]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.A.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Kosho, T.; Okamoto, N.; Ohashi, H.; Tsurusaki, Y.; Imai, Y.; Hibi-Ko, Y.; Kawame, H.; Homma, T.; Tanabe, S.; Kato, M.; et al. Clinical correlations of mutations affecting six components of the SWI/SNF complex: Detailed description of 21 patients and a review of the literature. Am. J. Med. Genet. Part A 2013, 161, 1221–1237. [Google Scholar] [CrossRef] [PubMed]
- Tsurusaki, Y.; Okamoto, N.; Ohashi, H.; Kosho, T.; Imai, Y.; Hibi-Ko, Y.; Kaname, T.; Naritomi, K.; Kawame, H.; Wakui, K.; et al. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat. Genet. 2012, 44, 376–378. [Google Scholar] [CrossRef]
- Ronan, J.L.; Wu, W.; Crabtree, G.R. From neural development to cognition: Unexpected roles for chromatin. Nat. Rev. Genet. 2013, 14, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Santen, G.W.E.; Aten, E.; Sun, Y.; Almomani, R.; Gilissen, C.; Nielsen, M.; Kant, S.G.; Snoeck, I.N.; Peeters, E.A.J.; Hilhorst-Hofstee, Y.; et al. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat. Genet. 2012, 44, 379–380. [Google Scholar] [CrossRef]
- Van Houdt, J.K.J.; Nowakowska, B.A.; Sousa, S.B.; Van Schaik, B.D.C.; Seuntjens, E.; Avonce, N.; Sifrim, A.; Abdul-Rahman, O.A.; Boogaard, M.-J.H.V.D.; Bottani, A.; et al. Heterozygous missense mutations in SMARCA2 cause Ni-colaides-Baraitser syndrome. Nat. Genet. 2012, 44, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Kathiriya, I.S.; Rao, K.S.; Iacono, G.; Devine, W.P.; Blair, A.P.; Hota, S.K.; Lai, M.H.; Garay, B.I.; Thomas, R.; Gong, H.Z.; et al. Modeling Human TBX5 Haploinsufficiency Predicts Regulatory Networks for Congenital Heart Disease. Dev. Cell 2021, 56, 292–309. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Yeo, G.; Muotri, A.R.; Kuwabara, T.; Gage, F.H. Noncoding RNAs in the mammalian central nervous system. Annu. Rev. Neurosci. 2006, 29, 77–103. [Google Scholar] [CrossRef] [Green Version]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Tsai, M.-C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [Green Version]
- Katz, M.G.; Fargnoli, A.S.; Kendle, A.P.; Hajjar, R.J.; Bridges, C.R. The role of microRNAs in cardiac development and regenerative capacity. Am. J. Physiol. Circ. Physiol. 2016, 310, H528–H541. [Google Scholar] [CrossRef] [Green Version]
- Takaya, T.; Nishi, H.; Horie, T.; Ono, K.; Hasegawa, K. Roles of MicroRNAs and Myocardial Cell Differentiation. In Genetics of Stem Cells: Part A; Progress in Molecular Biology and Translational Science; Academic Press: Cambridge, MA, USA, 2012; Volume 111, pp. 139–152. [Google Scholar] [CrossRef]
- Chen, J.-F.; Murchison, E.P.; Tang, R.; Callis, T.E.; Tatsuguchi, M.; Deng, Z.; Rojas, M.; Hammond, S.M.; Schneider, M.D.; Selzman, C.H.; et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc. Natl. Acad. Sci. USA 2008, 105, 2111–2116. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.-Z. The Role of MicroRNA-1 and MicroRNA-133 in Skeletal Muscle Proliferation and Differentiation. Nat. Genet. 2005, 38, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Samal, E.; Srivastava, D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 2005, 436, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Williams, A.H.; Kim, Y.; McAnally, J.; Bezprozvannaya, S.; Sutherland, L.B.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. An intragenic MEF2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proc. Natl. Acad. Sci. USA 2007, 104, 20844–20849. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; Von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of Cardiogenesis, Cardiac Conduction, and Cell Cycle in Mice Lacking miRNA-1-2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Bezprozvannaya, S.; Williams, A.H.; Qi, X.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. microRNA-133a reg-ulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008, 22, 3242–3254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wystub, K.; Besser, J.; Bachmann, A.; Boettger, T.; Braun, T. miR-1/133a Clusters Cooperatively Specify the Cardiomyo-genic Lineage by Adjustment of Myocardin Levels during Embryonic Heart Development. PLoS Genet. 2013, 9, e1003793. [Google Scholar] [CrossRef]
- Grunert, M.; Appelt, S.; Dunkel, I.; Berger, F.; Sperling, S.R. Altered microRNA and target gene expression related to Tetralogy of Fallot. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Tatsuguchi, M.; Seok, H.Y.; Callis, T.E.; Thomson, J.M.; Chen, J.-F.; Newman, M.; Rojas, M.; Hammond, S.M.; Wang, D.-Z. Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J. Mol. Cell. Cardiol. 2007, 42, 1137–1141. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Sun, Y.; Shan, H.; Li, X.; Zhang, M.; Zhou, X.; Xing, S.; Sun, H.; Chu, W.; Qiao, G.; et al. Beta-adrenoceptor regulates miRNA expression in rat heart. Med Sci Monit. 2012, 18, BR309. [Google Scholar]
- Miquerol, L.; Dupays, L.; Théveniau-Ruissy, M.; Alcoléa, S.; Jarry-Guichard, T.; Abran, P.; Gros, D. Gap Junctional Connexins in the Developing Mouse Cardiac Conduction System: Novartis Foundation Symposium; John Wiley & Sons: Hoboken, NJ, USA, 2003; Volume 250, pp. 80–109. [Google Scholar] [CrossRef]
- Huang, G.Y.; Cooper, E.S.; Waldo, K.; Kirby, M.L.; Gilula, N.B.; Lo, C.W. Gap Junction–mediated Cell–Cell Communication Modulates Mouse Neural Crest Migration. J. Cell Biol. 1998, 143, 1725–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.-M.; Zhang, K.; Li, Y.; Shi, K.; Liu, Y.-L.; Yang, Y.-F.; Fang, Y.; Mao, M. Screening miRNA and their target genes related to tetralogy of Fallot with microarray. Cardiol. Young 2013, 24, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Xu, X.; Deng, F.; Feng, J.; Zhang, H.; Liu, Y.; Zhang, Y.; Pan, L.; Liu, Y.; Zhang, D.; et al. mi RNA -940 reduction contributes to human Tetralogy of Fallot development. J. Cell. Mol. Med. 2014, 18, 1830–1839. [Google Scholar] [CrossRef]
- Hornstein, E.; Mansfield, J.H.; Yekta, S.; Hu, J.K.-H.; Harfe, B.D.; McManus, M.T.; Baskerville, S.; Bartel, D.P.; Tabin, C.J. The microRNA miR-196 acts upstream of Hoxb8 and Shh in limb development. Nature 2005, 438, 671–674. [Google Scholar] [CrossRef]
- Goddeeris, M.M.; Schwartz, R.; Klingensmith, J.; Meyers, E.N. Independent requirements for Hedgehog signaling by both the anterior heart field and neural crest cells for outflow tract development. Development 2007, 134, 1593–1604. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Chen, J.; Tian, T.; Zhou, X.; Gu, H.; Xu, L.; Zeng, Y.; Miao, R.; Jin, G.; Shen, H.; et al. Genetic variants of miRNA sequences and non-small cell lung cancer survival. J. Clin. Investig. 2008, 118, 2600–2608. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.-B.; Mei, J.; Jiang, L.-Y.; Jiang, Z.-L.; Liu, H.; Zhang, J.-W.; Ding, F.-B. MiR-196a2 rs11614913 T>C Polymorphism is Associated with an Increased Risk of Tetralogy of Fallot in a Chinese Population. Acta Cardiol. Sin. 2015, 31, 18–23. [Google Scholar] [CrossRef]
- Yu, K.; Ji, Y.; Wang, H.; Xuan, Q.; Li, B.; Xiao, J.; Sun, W.; Kong, X. Association of miR-196a2, miR-27a, and miR-499 polymorphisms with isolated congenital heart disease in a Chinese population. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef]
- Wen, H.; Zhang, R.; Li, Y.; Qian, H.; Yan, Z.; Chen, Y.; Li, G. Association between functional polymorphisms in the promoter of the miR-143/145 cluster and risk of conotruncal heart defects. Personal. Med. 2019, 16, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Morton, S.U.; Scherz, P.J.; Cordes, K.R.; Ivey, K.N.; Stainier, D.Y.R.; Srivastava, D. microRNA-138 modulates cardiac patterning during embryonic development. Proc. Natl. Acad. Sci. USA 2008, 105, 17830–17835. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Yi, K.; Yu, H.; Ding, Y.; Li, D.; Wei, Y.; You, T.; Xie, X. Correlation between pri-miR-124 (rs531564) polymorphism and congenital heart disease susceptibility in Chinese population at two different altitudes: A case-control and in silico study. Environ. Sci. Pollut. Res. 2019, 26, 21983–21992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sucharov, C.C.; Sucharov, J.; Karimpour-Fard, A.; Nunley, K.; Stauffer, B.L.; Miyamoto, S.D. Micro-RNA Expression in Hypoplastic Left Heart Syndrome. J. Card. Fail. 2015, 21, 83–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, F.; Wagner, R.A.; Wilson, K.D.; Xie, X.; Fu, J.-D.; Drukker, M.; Lee, A.; Li, R.A.; Gambhir, S.S.; Weissman, I.L.; et al. Transcriptional and Functional Profiling of Human Embryonic Stem Cell-Derived Cardiomyocytes. PLoS ONE 2008, 3, e3474. [Google Scholar] [CrossRef] [Green Version]
- Oksenberg, N.; Ahituv, N. The role of AUTS2 in neurodevelopment and human evolution. Trends Genet. 2013, 29, 600–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magome, T.; Hattori, T.; Taniguchi, M.; Ishikawa, T.; Miyata, S.; Yamada, K.; Takamura, H.; Matsuzaki, S.; Ito, A.; Tohyama, M.; et al. XLMR protein related to neurite extension (Xpn/KIAA2022) regulates cell–cell and cell–matrix adhesion and migration. Neurochem. Int. 2013, 63, 561–569. [Google Scholar] [CrossRef]
- Linask, K.K.; Manisastry, S.; Han, M. Cross Talk between Cell–Cell and Cell–Matrix Adhesion Signaling Pathways during Heart Organogenesis: Implications for Cardiac Birth Defects. Microsc. Microanal. 2005, 11, 200–208. [Google Scholar] [CrossRef]
- Kratsios, P.; Catela, C.; Salimova, E.; Huth, M.; Berno, V.; Rosenthal, N.; Mourkioti, F. Distinct Roles for Cell-Autonomous Notch Signaling in Cardiomyocytes of the Embryonic and Adult Heart. Circ. Res. 2010, 106, 559–572. [Google Scholar] [CrossRef]
- Cheng, Z.; Lib, L.; Li, Z.; Liu, M.; Yan, J.; Wang, B.; Ma, X. Two novel HAND1 mutations in Chinese patients with ventricular septal defect. Clin. Chim. Acta 2012, 413, 675–677. [Google Scholar] [CrossRef]
- Zhou, B.; Ma, Q.; Kong, S.W.; Hu, Y.; Campbell, P.H.; McGowan, F.X.; Ackerman, K.G.; Wu, B.; Tevosian, S.G.; Pu, W.T. Fog2 is critical for cardiac function and maintenance of coronary vasculature in the adult mouse heart. J. Clin. Investig. 2009, 119, 1462–1476. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.A.; Weinmann, A.S. Common themes emerge in the transcriptional control of T helper and developmental cell fate decisions regulated by the T-box, GATA and ROR families. Immunology 2009, 126, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Jongsma, H.J.; Wilders, R. Gap Junctions in Cardiovascular Disease. Circ. Res. 2000, 86, 1193–1197. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Chaboissier, M.-C.; Behringer, R.R.; Rowitch, D.H.; Schedl, A.; Epstein, J.A.; De Crombrugghe, B. Essential role of Sox9 in the pathway that controls formation of cardiac valves and septa. Proc. Natl. Acad. Sci. USA 2004, 101, 6502–6507. [Google Scholar] [CrossRef] [Green Version]
- Kurian, L.; Aguirre, A.; Sancho-Martinez, I.; Benner, C.; Hishida, T.; Nguyen, T.B.; Reddy, P.; Nivet, E.; Krause, M.N.; Nelles, D.A.; et al. Identification of Novel Long Noncoding RNAs Underlying Vertebrate Cardiovascular Development. Circulation 2015, 131, 1278–1290. [Google Scholar] [CrossRef]
- Klattenhoff, C.A.; Scheuermann, J.C.; Surface, L.E.; Bradley, R.K.; Fields, P.A.; Steinhauser, M.L.; Ding, H.; Butty, V.L.; Torrey, L.; Haas, S.; et al. Braveheart, a Long Noncoding RNA Required for Cardiovascular Lineage Commitment. Cell 2013, 152, 570–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, J.; Long, H.; Zhou, C.; Zheng, S.; Wu, H.; Guo, T.; Wu, Q.; Zhong, T.; Wang, T. Long noncoding RNA Braveheart promotes cardiogenic differentiation of mesenchymal stem cells in vitro. Stem Cell Res. Ther. 2017, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ounzain, S.; Micheletti, R.; Arnan, C.; Plaisance, I.; Cecchi, D.; Schroen, B.; Reverter, F.; Alexanian, M.; Gonzales, C.; Ng, S.Y.; et al. CARMEN, a human super enhancer-associated long noncoding RNA controlling cardiac specification, differentiation and homeostasis. J. Mol. Cell. Cardiol. 2015, 89, 98–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grote, P.; Herrmann, B.G. The long non-coding RNA Fendrr links epigenetic control mechanisms to gene regulatory networks in mammalian embryogenesis. RNA Biol. 2013, 10, 1579–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grote, P.; Wittler, L.; Hendrix, D.; Koch, F.; Währisch, S.; Beisaw, A.; Macura, K.; Bläss, G.; Kellis, M.; Werber, M.; et al. The Tissue-Specific lncRNA Fendrr Is an Essential Regulator of Heart and Body Wall Development in the Mouse. Dev. Cell 2013, 24, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.M.; Anderson, D.M.; McAnally, J.R.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Transcription of the non-coding RNA upperhand controls Hand2 expression and heart development. Nature 2016, 539, 433–436. [Google Scholar] [CrossRef] [Green Version]
- Ruan, J.; Zheng, L.; Hu, N.; Guan, G.; Chen, J.; Zhou, X.; Li, M. Long noncoding RNA SNHG6 promotes osteosarcoma cell proliferation through regulating p21 and KLF2. Arch. Biochem. Biophys. 2018, 646, 128–136. [Google Scholar] [CrossRef]
- Chang, L.; Yuan, Y.; Li, C.; Guo, T.; Qi, H.; Xiao, Y.; Dong, X.; Liu, Z.; Liu, Q. Upregulation of SNHG6 regulates ZEB1 expression by competitively binding miR-101-3p and interacting with UPF1 in hepatocellular carcinoma. Cancer Lett. 2016, 383, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Tian, J.; Shi, W.; Xia, H.; Zhu, Y. LncRNA SNHG6 is Associated with Poor Prognosis of Gastric Cancer and Promotes Cell Proliferation and EMT through Epigenetically Silencing p27 and Sponging miR-101-3p. Cell. Physiol. Biochem. 2017, 42, 999–1012. [Google Scholar] [CrossRef] [PubMed]
- Birgani, M.T.; Hajjari, M.; Shahrisa, A.; Khoshnevisan, A.; Shoja, Z.; Motahari, P.; Farhangi, B. Long Non-Coding RNA SNHG6 as a Potential Biomarker for Hepatocellular Carcinoma. Pathol. Oncol. Res. 2017, 24, 329–337. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhuang, J.; Lin, Y.; Wang, X.; Chen, J.; Han, F. Long noncoding RNA SNHG6 contributes to ventricular septal defect formation via negative regulation of miR-101 and activation of Wnt/β-catenin pathway. Die Pharmazie 2019, 74, 23–28. [Google Scholar]
- Buikema, J.W.; Zwetsloot, P.-P.M.; Doevendans, P.A.; Domian, I.J.; Sluijter, J.P.G. Wnt/β-Catenin Signaling during Cardiac Development and Repair. J. Cardiovasc. Dev. Dis. 2014, 1, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Pahnke, A.; Conant, G.; Huyer, L.D.; Zhao, Y.; Feric, N.; Radisic, M. The role of Wnt regulation in heart development, cardiac repair and disease: A tissue engineering perspective. Biochem. Biophys. Res. Commun. 2016, 473, 698–703. [Google Scholar] [CrossRef] [Green Version]
- Song, G.; Shen, Y.; Zhu, J.; Liu, H.; Liu, M.; Shen, Y.-Q.; Zhu, S.; Kong, X.; Yu, Z.; Qian, L. Integrated Analysis of Dysregulated lncRNA Expression in Fetal Cardiac Tissues with Ventricular Septal Defect. PLoS ONE 2013, 8, e77492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Zhu, W.; Zhang, B.; Wu, Y.; Yan, S.; Yuan, Y.; Zhang, H.; Li, J.; Sun, K.; Wang, H.; et al. The MALAT1 gene polymorphism and its relationship with the onset of congenital heart disease in Chinese. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Gu, M.; Zheng, A.; Tu, W.; Zhao, J.; Li, L.; Li, M.; Han, S.; Hu, X.; Zhu, J.; Pan, Y.; et al. Circulating LncRNAs as Novel, Non-Invasive Biomarkers for Prenatal Detection of Fetal Congenital Heart Defects. Cell. Physiol. Biochem. 2016, 38, 1459–1471. [Google Scholar] [CrossRef]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs Are the Predominant Transcript Isoform from Hundreds of Human Genes in Diverse Cell Types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef] [Green Version]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2012, 19, 141–157. [Google Scholar] [CrossRef] [Green Version]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Vicens, Q.; Westhof, E. Biogenesis of Circular RNAs. Cell 2014, 159, 13–14. [Google Scholar] [CrossRef] [Green Version]
- Lim, T.B.; Lavenniah, A.; Foo, R.S.-Y. Circles in the heart and cardiovascular system. Cardiovasc. Res. 2019, 116, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, J.; Liu, H.; Yin, J.; Zhang, M.; Yu, Z.; Miao, H. Circulating plasma circular RNAs as novel diagnostic biomarkers for congenital heart disease in children. J. Clin. Lab. Anal. 2019, 33, e22998. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Beketaev, I. Sumoylation in gene regulation and cardiac disease: Potential for drug discovery. Adv. Genomics Genet. 2014, 185. [Google Scholar] [CrossRef] [Green Version]
- Shetty, P.M.V.; Rangrez, A.Y.; Frey, N. SUMO proteins in the cardiovascular system: Friend or foe? J. Biomed. Sci. 2020, 27, 1–14. [Google Scholar] [CrossRef]
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Chaanine, A.H.; Kizana, E.; Park, W.J.; Hajjar, R.J. SUMO1-dependent modulation of SERCA2a in heart failure. Nature 2011, 477, 601–605. [Google Scholar] [CrossRef]
- Wang, J.; Chen, L.; Wen, S.; Zhu, H.; Yu, W.; Moskowitz, I.P.; Shaw, G.M.; Finnell, R.H.; Schwartz, R.J. Defective sumoylation pathway directs congenital heart disease. Birth Defects Res. Part A Clin. Mol. Teratol. 2011, 91, 468–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komal, S.; Zhang, L.-R.; Han, S.-N. Potential regulatory role of epigenetic RNA methylation in cardiovascular diseases. Biomed. Pharmacother. 2021, 137, 111376. [Google Scholar] [CrossRef]
- Wu, B.; Su, S.; Patil, D.P.; Liu, H.; Gan, J.; Jaffrey, S.R.; Ma, J. Molecular basis for the specific and multivariant recognitions of RNA substrates by human hnRNP A2/B1. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Tang, P.L.; Wang, J.; Bao, S.; Shieh, J.T.; Leung, A.W.; Zhang, Z.; Gao, F.; Wong, S.Y.; Hui, A.L.; et al. Mutations in Hnrnpa1 cause congenital heart defects. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N6-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yang, Y.; Sun, B.-F.; Chen, Y.-S.; Xu, J.-W.; Lai, W.-Y.; Li, A.; Wang, X.; Bhattarai, D.P.; Xiao, W.; et al. 5-methylcytosine promotes mRNA export—NSUN2 as the methyltransferase and ALYREF as an m5C reader. Cell Res. 2017, 27, 606–625. [Google Scholar] [CrossRef] [Green Version]
- Fahiminiya, S.; Almuriekhi, M.; Nawaz, Z.; Staffa, A.; Lepage, P.; Ali, R.; Hashim, L.; Schwartzentruber, J.; Abu Khadija, K.; Zaineddin, S.; et al. Whole exome sequencing unravels disease-causing genes in consanguineous families in Qatar. Clin. Genet. 2013, 86, 134–141. [Google Scholar] [CrossRef]
- Israeli, Y.; Gabalski, M.; Ball, K.; Wasserman, A.; Zou, J.; Ni, G.; Zhou, C.; Aguirre, A. Generation of Heart Organoids Modeling Early Human Cardiac Development Under Defined Conditions. SSRN Electron. J. 2020. [Google Scholar] [CrossRef]
- Rossi, G.; Broguiere, N.; Miyamoto, M.; Boni, A.; Guiet, R.; Girgin, M.; Kelly, R.G.; Kwon, C.; Lutolf, M.P. Capturing Cardiogenesis in Gastruloids. Cell Stem Cell 2021, 28, 230–240. [Google Scholar] [CrossRef]
- Li, X.; Martinez-Fernandez, A.; Hartjes, K.A.; Kocher, J.-P.A.; Olson, T.M.; Terzic, A.; Nelson, T.J. Transcriptional atlas of cardiogenesis maps congenital heart disease interactome. Physiol. Genom. 2014, 46, 482–495. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Syndrome | Genes | Loci | Cardiac Disease | % CHD |
|---|---|---|---|---|
| Alagille | JAG 1 NOTCH2 | 20p12.2 1p12-p11 | PPS, TOF, PA | >90 |
| CFC | BRAF KRAS MAP2K1 MAP2K2 | 7q34 12p12.1 15q22.31 19p13.3 | PVS, ASD, HCM | 75 |
| Cantu | ABCC9 | 12p12.1 | PDA, BAV, HCM, CoA, PE, AS | 75 |
| Char | TFAP2B | 6p12.3 | PDA, VSD | 58 |
| CHARGE | CHD7 | 8q12 | TOF, PDA, DORV, AVSD, VSD | 75–85 |
| Costello | HRAS | 11p15.5 | PVS, ASD, VSD, HCM, arrhythmias | 44–52 |
| DiGeorge | TBX1 | 22q11.2 deletion | Conotruncal defects, VSD, IAA, ASD, VR | 74–85 |
| Ellis-van Creveld | EVC EVC2 | 4p16.2 4p16.2 | Common atrium | 60 |
| Holt-Oram | TBX5 | 12q24.1 | VSD, ASD, AVSD, conduction defects | 50 |
| Kabuki | KMT2D KDM6A | 12q13 Xp11.3 | CoA, BAV, VSD, TOF, TGA, HLHS | 50 |
| Noonan | PTPN11 SOS1 RAF1 KRAS NRAS RIT1 SHOC2 SOS2 BRAF | 12q24.13 2p22.1 3p25.2 12p12.1 1p13.2 1q22 10q25.2 14q21.3 7q34 | Dysplastic PVS, ASD, TOF, AVSD, HCM, VSD, PDA | 75 |
| Williams-Beuren | 7q11.23 deletion (ELN) | 7q11.23 | SVAS, PAS, VSD, ASD | 80 |
| Carpenter | RAB23 | 6p11.2 | VSD, ASD, PDA, PS, TOF, TGA | 50 |
| Coffin-Siris | ARID1B SMARCB1 ARID1A SMARCB1 SMARCA4 SMARCE1 | 6q25 22q11 1p36.1 22q11.23 19p13.2 17q21.2 | ASD, AVSD, VSD, MR, PDA, PS, DEX, AS | 20–44 |
| Cornelia de Lange | NIPBL SMC1L1 SMC3 | 5p13 Xp11.22 10q25 | PVS, VSD, ASD, PDA | 33 |
| Mowat-Wilson | ZEB2 | 2q22.3 | VSD, CoA, ASD, PDA, PAS | 54 |
| Rubinstein-Taybi | CBP EP300 | 16p13.3 22q13.2 | PDA, VSD, ASD, HLHS, BAV | 33 |
| Smith-Lemli-Opitz | DHCR7 | 11q12-13 | AVSD, HLHS, ASD, PDA, VSD | 50 |
| Cyanotic CHD | Brief Description |
|---|---|
| Tetralogy of Fallot (TOF) | A common cyanotic CHD; characterized by pulmonary stenosis/right ventricular outflow tract obstruction, VSD, over-riding aorta and hypertrophy of the right ventricle |
| Transposition of the great arteries (TGA) | Discordant ventriculoarterial connection—the right ventricle is connected to the aorta (instead of pulmonary artery), and left ventricle to pulmonary artery (instead of aorta) |
| Double outlet right ventricle (DORV) | Both the aorta and pulmonary artery arise predominantly or completely, from the right ventricle |
| Persistent truncus arteriosus | Failure of septation of the primitive truncus into the aorta and pulmonary artery, resulting in a single, common arterial trunk that overlies a large VSD |
| Hypoplastic left heart syndrome (HLHS) | Underdevelopment of the left-sided structures of the heart, including the ascending aorta, left ventricle and aortic and mitral valves |
| DNA Methylation | |||||
|---|---|---|---|---|---|
| Allele | Clinical Sample Size | Modification | Tissue Type | Cardiac Disease Phenotype | Reference |
| NOX5 | 21 VSD and 15 controls | Hypermethylation | Fetal myocardial tissue | VSD | [78] |
| KIAA0310; RAB43; NDRG2 | 21 VSD and 15 controls | Hypermethylation | Fetal myocardial tissue | VSD | [79] |
| SIVA1 | Hypomethylation | ||||
| LINE-1* | 32 TOF and 15 controls [80]; 48 TOF patients and 16 controls [81] | Hypomethylation | Right ventricular tissue samples [80]; Right ventricular outflow tracts [81] | TOF | [80] |
| NKX2.5; HAND1; EGFR; EVC2; TBX5; CFC1B | 30 TOF and 6 controls [82]; 41 TOF and 6 control [83] | Hypermethylation | Right ventricular myocardium tissues | TOF, HLHS | [82,83] |
| GATA4; MSX1 | 6 Down syndrome with CHD, 6 Down syndrome without CHD, 6 isolated heart malformations, and 4 control | Hypermethylation | Whole heart tissue | AVSD, VSD, CoA, TOF, LHH, HAA, DORV, VSD, TOF, MVA, AVA, PFO, TVS; RHH, TA; ADA; TAV | [84] |
| SCO2 | 8 TOF, 8 ventricular septal defect, and 4 control | Hypermethylation | Myocardial biopsies | TOF, VSD | [85] |
| ZFPM2 | 43 TOF and 6 controls | Hypermethylation | Right ventricular outflow tract | TOF | [86] |
| p16INK4a | 63 TOF and 75 controls | Hypermethylation | Whole blood | TOF | [87] |
| BRG1 | 24 CHD and 11 controls | Hypomethylation | Various cardiac tissues. | TOF, VSD, DCRV | [88] |
| MTHFR | 40 Down syndrome without CHD; 40 mothers of Down syndrome with CHD, and 40 age matched control mothers | Hypermethylation | Whole blood | AVSD; VSD, ASD; TOF | [89] |
| TBX20 | 23 TOF and 5 controls [90]; 42 TOF and 6 controls [91] | Hypomethylation | Right ventricular myocardial tissues | TOF | [90] |
| ZIC3; NR2F2 | Monozygotic twin pair discordant for DORV | Hypermethylation | Whole blood | DORV | [92] |
| NRG1 | 7 Down syndrome patients with CHD and 9 Down syndrome without CHD | Hypermethylation | Whole blood | Endocardial cushion-type | [93] |
| Histone Modification | |||||
|---|---|---|---|---|---|
| Allele | Clinical Sample Size | Modification | Source Type | Cardiac Disease Phenotype | Reference |
| WHSC1 | Case study | H3K36me3 | In vivo mouse models [94] | HLH; WHS | [94,95,96] |
| MLL2; CHD7; WDR5; KDM5A; KDM5B | 362 severe CHD cases and 264 controls | H3K4me | Whole blood | LVO; CTD | [97] |
| UBE2B; RNF20; USP44 | H2BK120 | CTD; HTX; LVO | |||
| SMAD2 | H3K27 | HTX | |||
| EBAF | 16 VSD and 16 normal fetuses at 22–28 weeks of gestation. | H4ac | Myocardial tissue | VSD | [98] |
| RNF20; RNF40; UBE2B | 2645 case trios and 1789 control trios. | H2Bub1 | Whole blood or sputum | Dextrocardia; RAI; TAPVR; CAVC; PA; L-TGA; HLHS; TOF, RAA | [99] |
| JMJD1C; RREB1; MINA; KDM7A | 89 severe CHD cases and 95 controls | H3K27/H3K9 | Whole blood | CTD | [100] |
| KAT2B | 400 Chinese Han | HAT | Whole blood | TOF, TA and TGA, VSD, AVSD and PDA | [101] |
| PRDM6 | 35 individuals and their extended kindreds | H3K9me2/H4K20me2 | Whole blood | N-PDA | [102] |
| KANSL1 | 253 diseased patients | H4K16ac | Whole blood | TOF | [103] |
| Non-Coding RNA | |||||
|---|---|---|---|---|---|
| Allele | Clinical Sample Size | Modification | Tissue Type | Cardiac Disease Phenotype | Reference |
| miR-196a (rs11614913 CC) | 1324 CHD and 1783 controls | Increased mature miR-196a expression | Whole blood | TOF, VSD; ASD | [104] |
| miR-1-1 | 28 VSD and 9 controls | Upregulates GJA1 and SOX9 | Heart tissue | VSD | [105] |
| miR-181c | Downregulates BMPR2 | VSD | |||
| miR-1, miR-206 | 30 TOF and 10 controls | Upregulates Cx43 | Myocardium tissue | TOF | [106] |
| let-7e-5p; miR-222-3p; miR-433 | 3 VSD and 3 controls | Downregulated | Blood plasma | VSD | [107] |
| miR-184 | 10 CHD and 10 controls | Downregulated | Right ventricular outflow tract | Cyanotic cardiac defects | [108] |
| miRNA-139-5p | 5 family individuals | (c.1784T > C) gain-of-function | Whole blood | ASDII | [109] |
| miR-518e, miR-518f, and miR-528a | 7 Down syndrome patients with AVSD and 22 Down syndrome patients without CHD | Downregulates AUTS2 | Down syndrome lymphoblastoid cell lines (GSE34457) | AVSD | [110] |
| miR-518a, miR-518e, miR-518f, and miR-96 | Downregulates KIAA2022 | ||||
| miR-138 (rs139365823) | 857 CHD and 938 controls | Upregulates miR-138 | Whole blood | VSD; ASD; TOF; PDA | [111] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, T.B.; Foo, S.Y.R.; Chen, C.K. The Role of Epigenetics in Congenital Heart Disease. Genes 2021, 12, 390. https://doi.org/10.3390/genes12030390
Lim TB, Foo SYR, Chen CK. The Role of Epigenetics in Congenital Heart Disease. Genes. 2021; 12(3):390. https://doi.org/10.3390/genes12030390
Chicago/Turabian StyleLim, Tingsen Benson, Sik Yin Roger Foo, and Ching Kit Chen. 2021. "The Role of Epigenetics in Congenital Heart Disease" Genes 12, no. 3: 390. https://doi.org/10.3390/genes12030390
APA StyleLim, T. B., Foo, S. Y. R., & Chen, C. K. (2021). The Role of Epigenetics in Congenital Heart Disease. Genes, 12(3), 390. https://doi.org/10.3390/genes12030390